UFOP - CETEC - UEMG
REDEMAT
R
EDET
EMÁTICA EME
NGENHARIA DEM
ATERIAISUFOP – CETEC – UEMG
Tese de Doutorado
"
Modelos efetivos para separação solvente-soluto
através de nanoestruturas:
teoria e simulações computacionais "
Autor: Cláudia Karina Barbosa de Vasconcelos
Orientador: Prof. Alan Barros de Oliveira
Cláudia Karina Barbosa de Vasconcelos
“Modelos efetivos para separação solvente-soluto através de
nanoestruturas: teoria e simulações computacionais”
Tese de Doutorado apresentada ao Programa de Pós-Graduação em Engenharia de Materiais da REDEMAT, como parte integrante dos re-quisitos para a obtenção do título de Doutora em Engenharia de Materiais.
Área de concentração: Simulação Computacional Orientador: Prof. Alan Barros de Oliveira
Catalogação: www.sisbin.ufop.br V331m Vasconcelos, Cláudia Karina Barbosa de.
Modelos efetivos para separação solvente-soluto através de nanoestruturas [manuscrito]: teoria e simulações computacionais / Cláudia Karina Barbosa de Vasconcelos. - 2016.
83f.: il.: color; grafs.
Orientador: Prof. Dr. Alan Barros de Oliveira.
Tese (Doutorado) - Universidade Federal de Ouro Preto. Instituto de Ciências Exatas e Biológicas. Departamento de Física. Rede Temática em Engenharia de Materiais.
Área de Concentração: Simulação Computacional.
1. Simulação por computador. 2. Dessalinização da agua. 3. Extração por solventes. 4. Água. I. Oliveira, Alan Barros de. II. Universidade Federal de Ouro Preto. III. Titulo.
À minha família. — COM CARINHO.
Agradecimentos
Aos meus pais, Cláudio e Marta, o meu maior agradecimento. Vocês, que sempre primaram pelos meus estudos, são, com toda a certeza, os maiores responsáveis por mais esta conquista.
Ao meu esposo McGlennon. Muito obrigda por aguentar meus (muitos!) momen-tos de ansiedade e estresse. Sem falar no seu jeito metódico para me ajudar a conferir cada linha errada nos programas! Este doutorado sem você teria sido bem mais difícil. Sua compreensão e incentivo foram fundamentais.
Aos meus filhos, Ana Clara e Caio, que nem sempre souberam entender a minha ausência nos muitos momentos até a conclusão desta tese. Vocês foram, sem sombra de dúvida, a minha maior motivação.
Ao meu orientador, Prof. Alan, pela incomparável dedicação. Pelas diversas ma-drugadas, feriados e fins de semana que trabalhamos e discutimos este trabalho. Sem a sua compreensão e humanidade eu nunca teria conseguido! Você, com certeza, é o meu exemplo de profissionalismo!
Aos professores do Departamento de Física que, direta ou indiretamente, contri-buíram para o desenvolvimento desta pesquisa. Em especial a Profa. Taíse e o Prof. Ronaldo, meu muito obrigada!
Aos meus amigos e colegas da PUC-Minas, pelo apoio e risadas.
À CAPES, pelo apoio financeiro.
“If I have seen farther than others, it is because I stood on the shoulders of giants.” — SIRISAACNEWTON
Sumário
Agradecimentos vi
Lista de Figuras xiv
Lista de Abreviaturas xv
Lista de Simbolos xvi
Resumo xvii
Abstract xviii
1 Introdução 1
2 Metodologia 7
2.1 Dinâmica Molecular . . . 7
2.1.1 Algorítmo base . . . 8
2.1.2 Algorítmo de integração: “Velocity-Verlet” . . . 10
2.1.3 Raio de corte e lista de vizinhos . . . 11
2.1.4 Condições de contorno . . . 11
2.1.5 Ensemble estatístico . . . 12
2.1.6 Convenção da imagem mínima . . . 13
2.1.7 Termostatização e barostatização . . . 14
Equações de movimento para o ensembleNVT. . . 14
2.1.8 Configuração inicial, minimização e equilibração . . . 15
2.2 Potenciais de interação . . . 15
2.2.1 Potencial de Lennard-Jones (LJ) . . . 16
2.2.2 Potenciais do tipo caroço atenuado (Core-softened) . . . 17
3 O Modelo 20 3.1 Quantidades de interesse . . . 24
4 Resultados e Discussões 26 4.1 Caso 1:Pistão 2 fixo . . . 26
4.1.1 Campo Elétrico Externo . . . 34
4.1.2 Funcionalização do poro . . . 36
4.2 Caso 2:Pistão 2 submetido a pressões externas . . . 38
4.3 Modelo Analítico . . . 42
5 Considerações Finais 49
Bibliografia 51
Lista de Figuras
2.1 (a) Interações de pares existentes num conjunto de sete partículas. (b) Interações que envolvem apenas uma das sete partículas . . . 8 2.2 Diagrama que apresenta a idéia principal da dinâmica molecular. . . 9 2.3 Caixa de simulação sob condições periódicas de contorno . . . 12 2.4 Caixa de simulação, sob condições periódicas de contorno com raio de
corte,rc. . . 13
2.5 Comportamento típico da energia potencial como função da distância e a representação dos parâmetrosσ,ǫpara o potencial Lennard-Jones. . . 16 2.6 Potenciais do tipo caroço atenuado descontínuos do tipo ombro (a) e do
tipo rampa(b). . . 18 2.7 Potencial isotrópico de ombro repulsivo desenvolvido por de Oliveira
e colaboradores e descrito pela Equação 2.21. Os parâmetros utilizados forama= 5, r0/σ= 0.7ec= 1. . . 18
3.1 Sistema construído para simular o processo de separação soluto-solvente. 21 3.2 Esquema representativo do processo de deionização capacitiva. Uma
so-lução contento íons atravessa dois eletrodos submetidos a uma diferença de potencial. Os cátions são atraídos para o eletrodo negativo e os ânions são atraídos para o eletrodo positivo. . . 23 3.3 Construção da membrana com poro central com a primeira camada de
átomos (borda) diferenciada. A alteração do potencial de interação da borda representa a funcionalização ou a passivação do poro. . . 24 3.4 Comportamento da energia potencial de interação em função da
distân-cia para o potendistân-cial de Buckingham descrito pela Equação 3.5 e pelos parâmetros indicados anteriormente para a interação da borda com as partículas de solvente,A=−1,ρ= 0,5eC = 0. . . 24
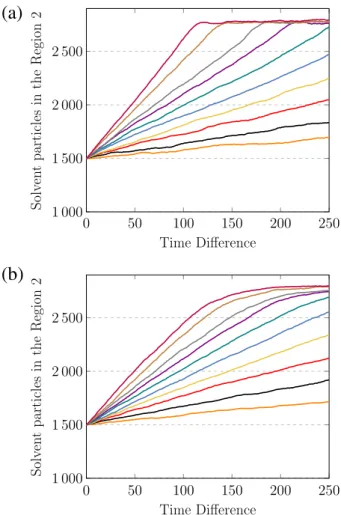
4.1 Número de partículas de solvente presentes na Região 2 como uma fun-ção do tempo. Cada curva corresponde a um valor diferente da pressão externa aplicada ao pistão 1, de baixo para cima: 1,0; 2,0; ...; 6,0; 8,0; 10,0; 12,0 e 14,0 em unidades de ǫ/σ3. Os resultados são apresentados para diferentes tamanhos de poros: (a) 14,2σ2 e (b)46,3σ2. A saturação observada nas curvas se deve ao tamanho finito do sistema, isto é, de-pois de um determinado período ocorre o esgotamento do reservatório de alimentação e o fluxo de solvente através do poro é interrompido. . . 27
4.2 (a) Resultados obtidos por Chen e Yang para o número de moléculas filtradas de água como uma função do tempo para uma membrana de grafeno para pressões entre 50 e 530 MPa. Os dados desta figura foram extraídos da referência [23]. (b) Dependência do número de moléculas filtradas de água com o tempo e com a área do poro observada por Hei-ranian e colaboradores para nanoporos deM oS2 a uma pressão de 250 MPa. Os dados desta figura foram extraídos da referência [50]. . . 28 4.3 Fluxo volumétrico de solvente como uma função do tempo. Cada curva
corresponde a um valor diferente da pressão externa aplicada ao pistão 1, de baixo para cima: 3,0;4,0;...; 9,0 em unidades deǫ/σ3. Os símbolos representam os dados simulados e as linhas são as regressões lineares destes dados. Os resultados são apresentados para diferentes tamanhos de poros: (a) 14,2σ2e (b) 46,3σ2. . . . 29 4.4 A quantidade de fluido por tempo (Φ) que flui num tubo de raio interno
Re comprimentoℓé proporcional às diferenças de pressão entre os dois lados, ∆P = P − P0 conforme o modelo de Hagen-Poiseuille. Esse modelo determina queΦ = D∆P, onde a constanteDdepende deR,ℓ e da viscosidadeηdo fluido. . . 29 4.5 Fluxo volumétrico de solvente como uma função da pressão aplicada
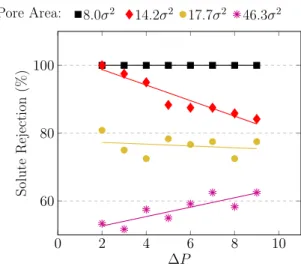
para diferentes áreas de poros. Os símbolos são dados simulados e li-nhas são as regressões lineares destes dados. . . 30 4.6 Rejeição percentual de soluto como uma função da pressão aplicada para
diferentes áreas de poro. Os símbolos são dados simulados e as linhas são as regressões lineares obtidas a partir desses dados. . . 31 4.7 Resultados apresentados por Zhu e colaboradores para a rejeição de sal
como uma função da pressão aplicada. Os dados apresentados nessa Figura foram extraídos da referência [21]. . . 31 4.8 Rejeição de sal, de acordo com Cohen-Tanugi e Jeffrey Grossman, em
função da pressão aplicada para poros passivados com (a) hidrogênios e (b) grupos hidroxilas. Os dados utilizados nesta Figura foram extraídos da referência [20]. . . 32 4.9 Comparação do fluxo volumétrico de solvente como função da pressão
aplicada para sistemas entre a Região 2 inicialmente vazia e a Região 2 inicialmente com 1500 partículas de solvente. Os símbolos representam os dados simulados e as linhas são as regressões lineares destes dados. Os resultados são apresentados para diferentes tamanhos de poros: (a) 14,2σ2e (b) 46,3σ2. . . . 33
4.10 Número de partículas de solvente que atravessaram a membrana como uma função do tempo para um sistema com a Região 2 inicialmente va-zia. Cada curva corresponde a um valor diferente da pressão externa aplicada ao pistão 1, de baixo para cima: 1,0; 2,0; ...; 6,0; 8,0; 10,0; 12,0 e 14,0 em unidades deǫ/σ3. Os resultados são apresentados para diferen-tes tamanhos de poros: (a) 14,2σ2e (b) 46,3σ2. . . . 34 4.11 Rejeição percentual de soluto como uma função da pressão aplicada para
diferentes áreas de poro. Os símbolos são dados simulados e as linhas são as regressões lineares obtidas a partir desses dados. A região 2 desse sistema inicialmente apresentava-se vazia. . . 34 4.12 Comportamento das partículas de soluto e solvente na Região 1 quando
submetidas a um campo elétrico (E = 3,0) paralelo aos pistões em fun-ção do tempo. Os íons positivos se acumulam nas proximidades do ele-trodo negativo enquanto que os íons negativos se concentram na região do eletrodo positivamente carregado. . . 35 4.13 Rejeição de soluto como função da pressão aplicada para sistemas com
campo elétrico externo com intensidades de 0,0; 1,0; 2,0 e 3,0 em unida-des reduzidas.Os símbolos representam os dados simulados enquanto que as linhas são as regressões lineares desses dados. Os resultados são apresentados para diferentes tamanhos de poros: (a) 14,2σ2 e (b) 46,3σ2. 36 4.14 Fluxo volumétrico de solvente como função da pressão aplicada para
sistemas com campo elétrico externo com intensidades de 0,0; 1,0; 2,0 e 3,0 em unidades reduzidas. Os símbolos representam os dados simula-dos e as linhas são as regressões lineares destes dasimula-dos. Os resultasimula-dos são apresentados para diferentes tamanhos de poros: (a) 14,2σ2 e (b) 46,3σ2. 36 4.15 Comparação da rejeição de soluto como função da pressão para
siste-mas com poro simples e com poro passivado. Os símbolos representam os dados simulados enquanto que as linhas são as regressões lineares desses dados. Os resultados são apresentados para diferentes tamanhos de poros: (a) 14,2σ2e (b) 46,3σ2. . . . 37 4.16 Comparação do fluxo volumétrico de solvente como função da pressão
para sistemas com poro simples e com poro passivado. Os símbolos representam os dados simulados e as linhas são as regressões lineares destes dados. Os resultados são apresentados para diferentes tamanhos de poros: (a) 14,2σ2e (b) 46,3σ2. . . . 37 4.17 Deslocamento do pistão como uma função do tempo para um sistema
com 1500 partículas de solvente. Os resultados são apresentados para as seguintes pressões externas, de baixo para cima, em unidades deǫ/σ3: 2,0; 2,1; 2,2; 2,3; 2,4; 2,5 e 2,6. . . 39
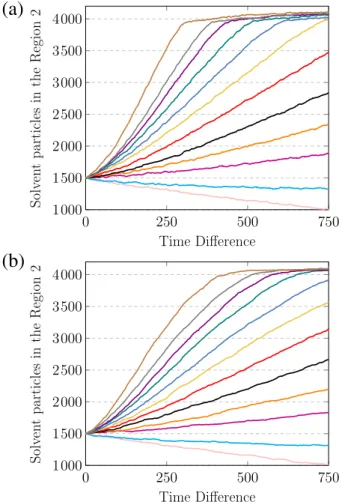
4.18 Número de partículas de solvente que atravessaram a membrana como uma função do tempo. Cada curva corresponde a um valor diferente de diferença de pressão ∆P = P1 −P2, de baixo para cima: -1,0; 0,0; ...; 6,0; 8,0; 10,0; 12,0 e 14,0 em unidades de ǫ/σ3. Os resultados são apresentados para diferentes tamanhos de poros: (a) 14,2σ2 e (b) 46,3σ2. 39 4.19 Resultados obtidos por Hu e colaboradores para dessalinização de água
através de uma membrana zeolítica, para o número de moléculas filtra-das de água como uma função do tempo de simulação. De baixo para cima∆P igual a 0, 30, 60, ...,150 MPa. Os dados utilizados nesta Figura foram extraídos da referência [19]. . . 40 4.20 A mesma curva mostrada na Figura 4.18(f) (∆P = 8,0ǫ/σ3) dividida em
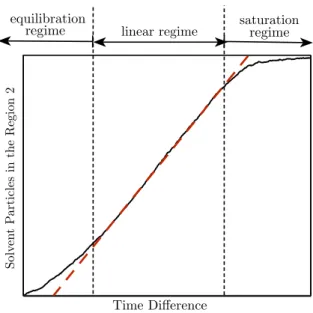
diferentes regimes em relação as partículas de solvente que atravessa-ram a membrana ao longo do tempo. Este resultado está de acordo com os resultados apresentados por Nicolai, Sumpter e Meunier para para a dessalinização da água através de membranas de óxido de grafeno [22]. 40 4.21 Fluxo volumétrico de solvente como uma função do tempo. Cada curva
corresponde a um valor diferente de diferença de pressão entre os dois pistões, de baixo para cima: 2,0; 3,0;...; 9,0 em unidades de ǫ/σ3. Os símbolos representam os dados simulados e as linhas são as regressões lineares destes dados. Os resultados são apresentados para diferentes tamanhos de poros:(a) 14,2σ2e (b) 46,3σ2. . . . 41 4.22 Fluxo volumétrico de solvente como uma função da diferença de pressão
aplicada entre os pistões para diferentes áreas de poros. Os símbolos são dados simulados e linhas são as regressões lineares destes dados. . . 41 4.23 Rejeição percentual de soluto como uma função da diferença de
pres-são entre os dois pistões para diferentes áreas de poro. Os símbolos pres-são dados simulados e as linhas são as regressões lineares obtidas a partir desses dados. . . 42 4.24 Número de partículas de solvente que atravessaram a membrana como
uma função do tempo. Cada curva corresponde a um valor diferente da pressão externa aplicada ao pistão 1, de baixo para cima: 1,0; 2,0; ...; 6,0; 8,0; 10,0; 12,0 e 14,0 em unidades de ǫ/σ3. Os resultados são apresentados para diferentes tamanhos de poros: (a) 14,2σ2e (b) 46,3σ2. As curvas foram divididas em duas regiões diferentes: regime transiente e regime permanente. A linha tracejada em preto representa o limite entre os dois regimes. . . 43 4.25 Esquema mostrandoN(t) para diferentes regimes de tempo. Quando
φA=µP, a curva é linear caracterizando a pressão crítica. Para pressões menores φA/µ, o comportamento observado é o subcrítico, enquanto que paraP > φA/µ, supercrítico. . . 45
4.26 Ajuste dos dados apresentados pela 4.24, com base na Equação 4.2. Os símbolos representam os dados simulados enquanto que as linhas são os ajustes baseados no modelo descrito. Cada curva corresponde a um valor diferente da pressão externa aplicada ao pistão 1, de baixo para cima: 1,0; 2,0; ...; 6,0; 8,0; 10,0; 12,0 e 14,0 em unidades deǫ/σ3. Os resul-tados são apresenresul-tados para diferentes tamanhos de poros: (a) 14,2σ2e (b) 46,3σ2. . . . 45 4.27 Número de partículas permeadas de soluto em função tempo de
simu-lação. Os símbolos representam os dados simulados enquanto que as linhas são os ajustes baseados no modelo descrito. Cada curva corres-ponde a um valor diferente da pressão externa aplicada ao pistão 1, de baixo para cima: 1,0; 2,0; ...; 6,0; 8,0; 10,0; 12,0 e 14,0 em unidades deǫ/σ3. Os resultados são apresentados para diferentes tamanhos de poros: (a) 14,2σ2e (b) 46,3σ2. . . . 46 4.28 Número de partículas filtradas de solvente em função tempo de
simula-ção para o sistema construído com 48.000 partículas de solvente e 1.960 mil partículas de soluto. Os símbolos representam os dados simulados enquanto que as linhas são os ajustes baseados no modelo. Cada curva corresponde a um valor diferente da pressão externa aplicada ao pistão 1, de baixo para cima: 1,0; 2,0; 3,0; 4,0; 6,0 e 8,0 em unidades deǫ/σ3. Os resultados são apresentados para diferentes tamanhos de poros: (a) 19,6σ2e (b) 38,5σ2. . . . 47 4.29 Número de partículas permeadas de soluto em função tempo de
simula-ção para o sistema construído com 48.000 partículas de solvente e 1.960 mil partículas de soluto. Os símbolos representam os dados simulados enquanto que as linhas são os ajustes baseados no modelo. Cada curva corresponde a um valor diferente da pressão externa aplicada ao pistão 1, de baixo para cima: 1,0; 2,0; 3,0; 4,0; 6,0 e 8,0 em unidades deǫ/σ3. Os resultados são apresentados para diferentes tamanhos de poros: (a) 19,6σ2e (b) 38,5σ2. . . . 47
Lista de Abreviaturas
DM Dinâmica molecular
DME Dinâmica molecular de equilíbrio
DMNE Dinâmica molecular de não equilíbrio
LJ Lennard-Jones
E Campo elétrico
Lista de Símbolos
N Partículas de solvente que atravessaram a membrana S Partículas de soluto que atravessaram a membrana ∆t Intervalo de tempo
P Pressão
φ Fluxo volumétrico de solvente
Π Pressão osmótica
S Partículas permeadas de soluto
UNIVERSIDADE FEDERAL DE OURO PRETO
Resumo
Departamento de Física
Rede Temática em Engenharia de Materiais
Doutorado em Engenharia de Materiais
Modelos efetivos para separação solvente-soluto através de nanoestruturas: teoria e simulações computacionais
por Cláudia Karina BarbosaDEVASCONCELOS
A escassez de água potável nos mais diversos países tem se mostrado um problema cada vez mais relevante. Estima-se que, atualmente, aproximadamente 748 milhões de pessoas no planeta não têm acesso à fontes de água potável. Sob esse aspecto, a des-salinização de água do mar tem se mostrado uma alternativa promissora, não apenas por97%da água do planeta estar concentrada nos oceanos e mares mas também pelo fato do percentual de água obtido desta maneira ainda ser muito pequeno. Em espe-cial, o processo de dessalinização da água através de nanoporos tem recebido grandes esforços científicos e tecnológicos. Apesar dos resultados animadores, um dos pro-blemas encontrados é que o processo de dessalinização de água é tipicamente macro. Mesmo com o poder computacional disponível atualmente, é impossível simular este problema em escala-macro. Daí a importância de se procurar modelos mais simples e computacionalmente mais baratos. Nesse sentido, uma alternativa eficaz para a si-mulação computacional de fluidos complexos é a utilização dos potenciais efetivos do tipo caroço atenuado (core-softened potentials). Estes potenciais tem sido utilizados para investigar fluidos anômalos dando bons resultados na descrição de propriedades di-nâmicas, termodinâmicas e estruturais desses fluidos. Devido à sua simplicidade, as simulações se tornam mais rápidas e tratamentos matemáticos se tornam possíveis. Baseado nesses potenciais, este trabalho propõe um modelo para a separação soluto solvente a partir de osmose reversa. O comportamento das partículas filtradas de sol-vente e as propriedades, tais como rejeição de soluto e fluxo volumétrico de solsol-vente, foram comparadas com resultados prévios apresentados na literatura para modelos moleculares clássicos. O objetivo deste trabalho é o de dar os primeiros passos para o desenvolvimento de um modelocoarse-grainedde dessalinização de água, onde a abor-dagem de problemas em escalas maiores, tanto em tempo quanto em tamanho, possam ser viáveis computacionalmente.
FEDERAL UNIVERSITY OF OURO PRETO
Abstract
Physics DepartmentREDEMAT
Materials Engineering (PhD)
An effective model for solvent - solute separation : molecular dynamics simulations
by Cláudia Karina BarbosaDEVASCONCELOS
Currently, the shortage of potable water in several countries has been an increasin-gly relevant problem. It is estimated that nowadays approximately 748 million people around the globe no longer have access to drinking water sources. Seawater desalina-tion has proven to be a promising alternative, not only because 97%of water on the planet is located in the oceans and seas, but also because the percentage of water ob-tained in this way is still very small. Especially water desalination through nanopores has received great scientific and technological efforts. Despite encouraging results, the water desalination problem is typically macro. Considering the modern computational power available, it is literally impossible to study this problem using an all-atoms ap-proach in a macro size scale. In this sense, it is important to seek for cheaper, alternative procedures to attack this problem. An alternative to computationally model water-like fluids in an effective manner relies on core-softened potentials. Such potentials have been used to investigate anomalous fluids giving good results in the description of dynamic, thermodynamic and structural properties of these fluids. Due to their sim-plicity, computational simulations become faster and mathematical treatments are pos-sible. This work propose an effective model for solute separation from fluids through reverse osmosis based on core-softened potentials. The behavior of solvent particles filtered and properties such as solute rejection and solvent volumetric flow rate were compared with previous results reported in the literature for classical molecular mo-dels.The purpose of this work is to pave the first steps towards coarse-grained models for water desalination processes which addressing problems at larger scales, both in time and in size, can be computationally feasible.
Capítulo 1
Introdução
A escassez hídrica é uma questão que tem sido incansavelmente discutida em todo o mundo. Estima-se que, atualmente, em torno de748milhões de pessoas no planeta não têm acesso a fontes de água potável. A previsão é que, em 2025, 48 países, com uma população total de aproximadamente 2,9 bilhões de pessoas, sofram com a falta d’água [1]. Um relatório divulgado pela ONU em 2015 afirma que, se nada for feito, o déficit de abastecimento pode chegar a 40% em 2030 [2]. Essas crises de abastecimento têm sido creditadas à seca, ao desperdício e ao excesso de consumo e, dentre as soluções mais discutidas, destacam-se o redirecionamento de rios, a proteção dos mananciais, a concientização da redução do consumo e o investimento em técnicas de potabiliza-ção. Nesse cenário, a dessalinização de água do mar tem se mostrado uma alternativa bastante promissora. De acordo com o relatório divulgado pela organização britânica Global Water Intelligence, no ano de 2009, a água dessalinizada respondeu por um au-mento de cerca de 10% da capacidade de produção global [3]. Este pequeno percentual [4] associado ao fato de 97% da água do planeta estar concentrada nos oceanos e mares, evidencia a importância do aprimoramento e investimento nessa técnica.
A dessalinização envolve processos de separação soluto-solvente e tem como ob-jetivo principal remover o sal que se encontra na água. Atualmente, as metodologias mais empregadas envolvem técnicas como destilação, osmose reversa (OR), dessalini-zação térmica e congelamento [4–9]. Todas elas têm um fator em comum: envolvem processos muito caros e pouco eficientes [4, 5, 10, 11]. Se comparados, os processos de separação por membranas são energeticamente mais favoráveis do que os processos térmicos e apresentam, ainda, uma maior flexibilidade operacional [12]. Nesse con-texto, a osmose reversa tem recebido grande atenção da comunidade científica. A OR tem, como força motriz, um gradiente de pressão e utiliza uma membrana seletiva e semipermeável que restringe a passagem de uma ou mais espécies químicas do reser-vatório de alimentação para o permeado. A aplicação de uma pressão externa maior do que a pressão osmótica do fluido força a passagem de solvente através da membrana. As membranas convencionais utilizadas para este fim são caras e pouco eficientes, o transporte de água é lento e há grandes dificuldades para se controlar a seletividade da membrana [13]. As pesquisas desenvolvidas nesta área visam, geralmente, reduzir o consumo de energia, aumentando a recuperação de água e diminuindo a diferença de pressão entre os reservatórios de alimentação e permeado [14–17].
Capítulo 1. Introdução 2
Com o advento das nanoestruturas, em especial das membranas nanoporosas de grafeno, os processos de filtragem e dessalinização de água têm obtido resultados ani-madores, onde a separação de água e sal através de nanoestruturas tem ganhado des-taque na comunidades científica. A exclusão por tamanho faz com que os nanoporos possam ser usados como filtros, isto é, em função da sua dimensão, permitem a passa-gem apenas das moléculas menores.
Um dos primeiros trabalhos que levou em conta a dessalinização de água e nano-estruturas foi desenvolvido por Yu-xiang Jia e colaboradores, em 2010 [18]. Os autores sugeriram a utilização de nanotubos de carbono como membranas no processo de des-salinização de água. As simulações mostraram que estas estruturas apresentam boa rejeição de sal, aliada a boas propriedades mecânicas e um fluxo de água razoável.
Em 2011, Zhongqiao Hu, Yifei Chen, and Jianwen Jiang estudaram uma estrutura organometálica e nanoporosa de imidazolato zeolítico (ZIF -8), como uma membrana de osmose reversa para purificação de água [19]. Segundo as simulações, o número de moléculas filtradas de água apresenta uma relação linear com o tempo e o sentido do fluxo de água está relacionado com a diferença de pressão entre os reservatórios de alimentação e do permeado.
Em 2012, Cohen-Tanugi e Grossman mostraram que poros nanométricos em mem-branas de grafeno podem separar cloreto de sódio e água com grande eficácia [20]. Com cálculos de dinâmica molecular, eles sugeriram que a capacidade da membrana para impedir a passagem de sal depende diretamente do tamanho, da funcionalização química do poro e da pressão externa aplicada.
No ano de 2013, Zhu e colaboradores investigaram a importância do tamanho do poro para a dessalinização através de folhas de grafino do tipo 3, 4, 5 e 6 [21]. A es-trutura do grafino é constituída por uma folha plana de átomos com muitas formas de conjugação entre os grupos acetileno e fenil. O tamanho de poro pode ser ajustado alterando o número de ligações de acetileno (definido como n) entre os anéis fenils adjacentes. O grafino-3 apresenta 3 ligações, para o grafino-4,n= 4, e assim sucessiva-mente. Em outras palavras, a principal diferença entre as estruturas por eles estudadas é o tamanho da porosidade efetiva para passagem de solução. Eles mostraram que para poros muito pequenos a rejeição é de 100%, independentemente da pressão ex-terna, devido à exclusão por tamanho, enquanto que para os poros maiores a rejeição diminui com o aumento da pressão externa aplicada. Para as áreas de poros maiores, foi registrado um leve aumento da rejeição com o mesmo aumento da pressão externa em função das barreiras de energia para a passagem de íons serem muito maiores do que a barreira encontrada pela água.
Capítulo 1. Introdução 3
maiores, em função do tamanho finito do sistema, i.e. o esgotamento do reservatório de alimentação, aparece o regime de saturação. O regime linear, por sua vez, surge para tempos intermediários [22].
Em 2015, Qi Chen e Xiaoning Yang investigaram, computacionalmente, o desempe-nho de nanoporos funcionalizados de grafeno como membrana de osmose reversa para dessalinização. Eles mostraram que pressões mais elevadas levam a um maior número de moléculas filtradas de água e que a passivação do poro tem papel importante na eficiência da membrana [23]. Ainda em 2015, Perreault e colaboradores fizeram uma revisão das aplicações ambientais de materiais a base de grafeno [24]. Esses materiais foram colocados como uma ferramenta importante para solução de desafios globais, tanto no desenvolvimento de novas tecnologias quanto no melhoramento das tecnolo-gias já existentes. Um dos pontos de destaque foi a utilização do grafeno como base da próxima geração de membranas para o tratamento de água. Além disso, o processo de deionização capacitiva foi indicado como um processo emergente e de grande po-tencial, uma vez que a exigência de energia desse processo pode ser menor do que para a tradicional osmose reversa. A deionização capacitiva baseia-se na remoção dos íons presentes em solução, armazenando-os na interface eletrodo-solução através da aplicação de uma diferença de potencial [25].
Mais recentemente, em 2016, David Coehn-Tanugi estudaram através de simula-ções clássicas de dinâmica molecular a eficiência de grafeno multicamadas como mem-brana para o processo de dessalinização de água [26]. Este trabalho estabelece um entendimento em nível atômico dos efeitos de camadas adicionais de grafeno nanopo-roso. Sob o ponto de vista experimental, filmes de grafeno multicamadas podem ser sintetizados a um custo muito inferior em relação ao grafeno em camada única.
Por outro lado, trabalhos envolvendo experimentos em que nanoestruturas são uti-lizadas como membranas seletivas para o processo de dessalinização de água ainda estão em fase inicial de desenvolvimento. A dificuldade e o alto custo envolvido na obtenção experimental, por exemplo, de poros em filmes de grafeno, que apresentem uma faixa restrita de tamanho (i.e.), em termos de área, poros capazes de bloquear con-sideravelmente a passagem de sal e permitir um bom fluxo de água) aumentam ainda mais os desafios tecnológicos envolvidos no processo de dessalinização de água.
Capítulo 1. Introdução 4
em que nanoporos foram criados por bombardeamento por íons Ga+ e elétrons produ-zindo membranas de grafeno com estabilidade química e mecânica e também flexíveis. Quando essas membranas foram utilizadas na dessalinização, uma taxa de rejeição de 100% foi alcançada, um resultado animador para a continuidade das pesquisas nessa área de conhecimento. Em 2016 Bhadra e colaboradores demostraram e eficiência da confecção de uma membrana por meio da imobilização de óxido de grafeno em poli-tetrafuoretileno (PTFE) para dessalinização [30]. A completa rejeição salina obtida foi justificada pelas características da membrana desenvolvida como a presença de grupos polares no GO, a sorção seletiva e o efeito de nanocapilaridade.
Nesse contexto, grandes esforços tem sido feitos afim de se compreender os prin-cipais mecanismos responsáveis pela dessalinização de água em uma escala nanomé-trica. Inúmeros outros trabalhos indicam que membranas baseadas em nanoestruturas são capazes de rejeitar os íons e aumentar o fluxo de água e a permeabilidade em várias ordens de grandeza, quando comparados com os processos de separação existentes [13, 31–50]. E, apesar dos resultados animadores, um dos problemas encontrados é que o processo de dessalinização de água é tipicamente macro. Mesmo com o poder compu-tacional disponível atualmente, é impossível simular este problema levando em conta todos os átomos existentes no sistema real.
O simples fato das simulações envolverem água já mostra o quão desafiador é o problema. Apesar da água ser considerada o líquido mais importante para a vida, ela ainda não é muito bem compreendida. Tal fato se deve, principalmente, à presença de diversas anomalias, tais como a anomalia estrutural, a anomalia na difusão e na den-sidade. Com o intuito de incluir esses efeitos nas simulações computacionais centenas de modelos moleculares foram propostos para água. Tais modelos fornecem, para a temperatura associada a máxima densidade, valores que variam de -45 (modelo SPC) até 25oC (modelo POL5/TZ), enquanto que o valor encontrado experimentalmente é de4oC a 1 atm. O modelo TIP5P foi concebido com o intiuito de se obter o valor de
4oC, mas falha em muitos outros aspectos. Apesar de reproduzirem razoavelmente bem o diagrama de fases da água, esses modelos são complicados e possuem um custo computacional elevado. Mesmo com as limitações envolvidas, esses modelos têm sido amplamente utilizados no estudo dos processos de dessalinização de água. As simu-lações de primeiros princípios seriam mais apropriadas para uma análise quantitativa, entretanto, para representar minimamente o problema, seriam necessários milhares de átomos e milhões de passos de simulação. Nesse sentido, na maioria dos casos, técnicas ab initiotornam-se impraticáveis.
Capítulo 1. Introdução 5
Para a água, por exemplo, os comportamentos anômalo termodinâmico, dinâmico e estrutural são reproduzidos com sucesso por esses potenciais. Um outro ponto im-portante é que não há direcionalidade e nem a presença de cargas. Esses ingredientes transformam os modelos efetivos em ferramentas importantes e de baixo custo com-putacional. Esses tipos de modelos têm sido fortemente explorados não só computa-cionalmente mas também por meio de ferramentas teóricas devido à sua simplicidade [51–62]. Os sucessivos bons resultados deram suporte ao uso dos potenciais efetivos em ambientes mais complexos, como, por exemplo, no confinamento de água entre placas [63–65].
Miha Luksic e colaboradores reproduziram computacionalmente uma solução sa-lina e estudaram como a presença de íons pode alterar o conjunto de anomalias es-truturais, termodinâmicas e dinâmicas de um fluido. O solvente foi simulado através de potenciais do tipo caroço atenuado, enquanto os íons positivos e negativos foram modelados como esferas rígidas carregadas [66]. O estudo permitiu, dentre outras con-clusões, a determinação das regiões que delimitam as anomalias estruturais e termodi-nâmicas para esse modelo de mistura.
Bordin et. al verificaram o efeito de confinamento dentro de nanotubos no com-portamento dinâmico de um fluido. Modelado por meio de um potencial efetivo o fluido foi capaz de reproduzir o comportamento anômalo observado na densidade e na difusão de água no estado líquido. Os resultados indicaram uma aumento no co-eficiente de difusão da água para nanotubos de pequenos raios [67]. A conexão entre as propriedades estruturais e dinâmicas do fluido confinado em nanoporos também foi discutida [68]. Ainda com esses potenciais, José Rafael Bordin, Alexandre Diehl e Marcia C. Barbosa estudaram o comportamento termodinâmico de um fluido anômalo confinado dentro de nanoporos. O sistema apresentou as anomalias de difusão e den-sidade observadas na água líquida, mostraram a formação de camadas em um fluido anômalo confinado dentro nanoporos e que a natureza do confinamento é relevante para as propriedades do líquido confinado [69].
É nesse cenário que este trabalho propõe a utilização de potenciais efetivos como base para a construção de um sistema simples de separação soluto-solvente. Esse mo-delo, mais barato computacionalmente, permitirá simulações com uma escala mais re-alista de partículas e deve ser capaz de reproduzir as principais características do pro-cesso de separação do sal da água apresentadas até então na literatura por meio de simulações envolvendo modelos moleculares clássicos. O objetivo deste trabalho é o de dar os primeiros passos para o desenvolvimento de um modelo coarse-grainedde dessalinização de água, onde a abordagem de problemas em escalas maiores, tanto em tempo quanto em tamanho, possam ser viáveis computacionalmente.
Capítulo 1. Introdução 6
Capítulo 2
Metodologia
Neste capítulo são apresentados os conceitos básicos da técnica de Dinâmica Molecular, do Potencial de Lennard-Jones e dos potenciais do tipo caroço atenuado.
2.1
Dinâmica Molecular
Para estudar a evolução de um sistema de partículas em função do tempo, na década de 1950, Alder e Wainright desenvolveram um método [70], hoje conhecido como Di-nâmica Molecular DM. Esse método foi aprimorado por Rahman [71] e acabou con-sagrado na área da física estatística computacional. Desde então, o método tem sido aplicado à modelagem de sistemas físicos que vão desde um fluido simples, como um gás nobre diluído [72], até sistemas biológicos complexos, como um vírus [73]. Con-siderando os recursos computacionais disponíveis atualmente, sistemas da ordem de 109 partículas já podem ser simulados através da DM [74].
O método resolve numericamente as equações clássicas de movimento para um conjunto de partículas que interagem entre si e, com isso, a trajetória no espaço de fa-ses do sistema ao longo do tempo é gerada. Em cada passo temporal, propriedades instantâneas do sistema podem ser calculadas. Por fim, é realizado o cálculo das pro-priedades macroscópicas de interesse.
Na literatura, a Dinâmica Molecular é apresentada sob dois aspectos: dinâmica mo-lecular de equilíbrio (DME) e dinâmica momo-lecular de não equilíbrio (DMNE). A DME permite o cálculo de propriedades a partir de flutuações do estado de equilíbrio. A DMNE baseia-se na aplicação de uma perturbação externa, claramente definida, tal qual uma deformação geométrica, ou pela imposição de um gradiente de temperaturas [75, 76] . Vale ressaltar que uma das vantagens da DMNE é que ela imprime à simula-ção uma maior semelhança com os procedimentos experimentais, no que se refere ao cálculo das propriedades de interesse [77].
Informações adicionais sobre o método podem ser encontradas em referências clás-sicas sobre o assunto [77–81].
Capítulo 2. Metodologia 8
2.1.1 Algorítmo base
Em uma simulação de dinâmica molecular, as equações clássicas de Newton para o movimento são resolvidas por integração numérica levando em conta um conjunto de partículas que interagem entre si, confinadas numa caixa de simulação virtual [77]. As equações do movimento para cada partículaAdo sistema envolvem um conjunto de equações diferenciais de primeira ordem:
vA(t) =r˙A(t) (2.1)
aA(t) =v˙A(t) (2.2)
FA(t) =maA(t) (2.3)
onde rA(t),vA(t), aA(t), FA(t) são, respectivamente, os vetores posição, velocidade,
aceleração e força resultante sobre a partículaA, no instantet.
A força resultante que atua sobre cada partículaAestá relacionada com o potencial de interação entre a partículaA e todas as demais partículasB do sistema. A Figura 2.1(a) considera as interações de pares existentes num conjunto de sete partículas. Já a Figura 2.1(b), por simplificação, mostra apenas as interações que envolvem apenas uma das sete partículas. Cada parAB define um potencialU. O gradiente desse po-tencial equivale a forçaf que a partículaB imprime sobre a partículaA[82]. A força resultante,FA, em cada instante de tempo, é a soma destas forças de interação, ou seja,
FA(t) = X
B6=A
f(t) = X
B6=A
−∇U(t). (2.4)
A B1
B2 B3
B4
B5
B6
B7
(a)
A B1
B2 B3
B4
B5
B6
B7
(b)
FIGURA2.1: (a) Interações de pares existentes num conjunto de sete partículas. (b)
Intera-ções que envolvem apenas uma das sete partículas
Capítulo 2. Metodologia 9
estejam acopladas. Isso implica na necessidade de se acumular informações a respeito de posição e velocidade, sobre cada instantet, afim de se calcular as mesmas grandezas no instante posteriort+ ∆t.
A Figura 2.2 apresenta um diagrama com a idéia principal da dinâmica molecu-lar. Inicialmente, define-se a distribuição inicial de partículas, estruturada ou aleatória, bem como suas posições, velocidades e potenciais de interação. Os potenciais de inte-ração, por sua vez, levam às forças de interação que são responsáveis, em parte, pelo movimento das partículas. A repetição deste processo define a evolução temporal do sistema modelado.
Partindo de uma distribuição inicial de partículas, são definidas as velocidades das partículas. As posições permitem calcular os potenciais de interação, que por sua vez nos levam às forças das interações. Essas forças estabelecem o movimento das par-tículas. A repetição deste processo define a evolução dinâmica temporal do sistema modelado.
Geração dasN
partículas do sistema
Posições e velocidades
originais
Cálculos das interações Cálculos
das novas posições e velocidades
t
t
+ ∆
t
Capítulo 2. Metodologia 10
2.1.2 Algorítmo de integração: “Velocity-Verlet”
Há na literatura uma grande variedade de métodos consagrados para a integração das equações de movimento utilizadas em Dinâmica Molecular [80]. Um dos métodos mais adotados atualmente é o algorítmo de integração Velocity-Verlet [83], uma variação do algoritmo tradicional[84]. Este método admite apenas uma escala de tempo para os fenômenos modelados e, além disso, conserva a energia total e a quantidade de movimento do sistema. Sua formulação pode ser obtida por uma sequência simples de manipulações algébricas.
Como a trajetória clássica da partícula é contínua, a posição da partículaA, deter-minada porrA, num instante de tempo (tn+ ∆t), pode ser aproximada por uma série
de Taylor considerando até o termo de segunda ordem:
rA(tn+ ∆t) =rA(tn) + ∆tvA(tn) +
1 2∆t
2a
A(tn) +O(∆t3) (2.5)
ondetné o instante de referência qualquer,∆to passo temporal evAeaA, a velocidade
e a aceleração da partícula A, respectivamente. Isto significa que a posição de uma partícula A no instante tn + ∆t pode ser obtida a partir de rA, vA e aA obtidos no
instantetn.
Para obter a velocidade num instante futuro, é necessário usar um artifício mate-mático. Calcula-sevA(tn+12∆t)de duas formas:
vA
tn+
1 2∆t
=vA(tn) +
1
2∆taA(tn) +O(∆t
2), (2.6)
vA
tn+
1 2∆t
=vA(tn+ ∆t)−
1
2∆taA(tn+ ∆t) +O(∆t
2). (2.7)
Então, a segunda equação (Eq. 2.7) é subtraída da primeira (Eq. 2.6):
0 =vA(tn)−vA(tn+ ∆t) +
1
2∆taA(tn) + 1
2∆taA(tn+ ∆t) +O(∆t
2), (2.8)
o que leva a
vA(tn+ ∆t) =vA(tn) +
1
2∆t[aA(tn+ ∆t) +aA(tn)] +O(∆t
2). (2.9)
Tanto na Eq. 2.5 como na Eq. 2.9, os valores deaA(tn) eaA(tn+ ∆t)podem ser
obtidos pela Segunda Lei de Newton, considerando-se as interações entre a partícula desejada e as partículas vizinhas por meio da seguinte expressão:
aA(t) =
1 mA
X
B6=A
f(r, tn). (2.10)
Isto torna possível a determinação de vA(tn+ ∆t) na Eq. 2.9, já que, nesta etapa do
Capítulo 2. Metodologia 11
2.1.3 Raio de corte e lista de vizinhos
A definição de um raio de corte, rc, para o potencial é uma das estratégias mais
co-muns para se reduzir o custo computacional de uma simulação, uma vez que a etapa de cálculo dos potenciais de interação entre pares não ligados é a etapa que mais con-some recursos. Dessa forma, acima desse raio, o potencial é truncado, e passa a ser considerado nulo [77, 80].
Essa estratégia, assim como outras semelhantes, inclui um fator de erro no cálculo da energia total dos pares. Caso a simulação leve em conta um número muito grande de partículas, o valor desse erro pode afetar significativamente os resultados. Para raios de corte pequenos, costuma-se empregar correções de longo alcance para o potencial [77]. Tais correções se baseiam no cálculo de um valor médio dessas energias, a partir do raio de corte até o infinito, e são somadas à energia total de pares calculada.
Outra estratégia usada para aumentar a eficiência computacional diz respeito à de-finição das partículas “vizinhas” com as quais o potencial de interação será calculado. Num primeiro momento, para cada passo temporal, deveria-se buscar as partículas vi-zinhas, cujas distâncias de referência fossem menores que o raio de corte. Essa busca realizada em cada passo permitiria estabelecer uma lista de partículas vizinhas. En-tretanto, algumas simplificações podem ser implementadas visando a diminuição do custo computacional. Em alguns casos, a lista de vizinhos não necessariamente precisa ser atualizada em cada passo temporal. Como consequência, passariam a ser calcula-das apenas as interações entre as partículas enumeracalcula-das, em cada lista de vizinhos, em intervalos de tempo definidos e múltiplos do passo temporal.
2.1.4 Condições de contorno
Capítulo 2. Metodologia 12
FIGURA2.3: Caixa de simulação sob condições periódicas de contorno
2.1.5 Ensemble estatístico
Um ensemble é, segundo a física matemática moderna, uma família de medidas de probabilidade invariantes, indexadas pelos parâmetros macroscópicos do sistema estu-dado, que satisfaz o Princípio de Boltzmann-Gibbs, ou seja, para toda função de estado “relevante”F, no espaço de fase, o correspondente valor macroscópico em equilíbrio é dado pelo valor esperado (ou valor médio) deFcom respeito a uma medida de proba-bilidade “adequada”P(quandoN→ ∞, sendoNo número de partículas do sistema) [88]:
F =hFi≡
Z
ΩF(ω)P(dω) (2.11)
ondeF é o valor em equilíbrio da grandeza macroscópica associada,ωé o microestado no espaço de fase eΩé o espaço de fase.
Um ensemble deve ser ortódico, ou seja, sob variações infinitesimais, os parâmetros devem satisfazer as relações usuais da termodinâmica do equilíbrio para o sistema macroscópico dado. Há vários tipos de ensembles demonstravelmente ortódicos (em alguns casos somente no limite termodinâmico) que podem tornar-se convenientes, dependendo das circunstâncias a que o sistema é submetido e do experimento que se quer reproduzir, são eles: o microcanônico ou NVE (número de partículas, volume e energia total constantes durante a simulação), canônico ou NVT (com número de partículas, volume e temperatura constantes) e o grande canônico ou µVT(potencial químico, volume e temperatura constantes) [88, 89].
Capítulo 2. Metodologia 13
cinética média das partículas e, consequentemente, com a velocidade média das partí-culas do sistema,
KBT =m
D
v2jE , (2.12)
ondemé a massa da partícula evj é a j-ésima componente da velocidade. O valor da
temperatura pode ser, então, controlado reescalando-se as velocidades.
2.1.6 Convenção da imagem mínima
A implementação das condições periódicas de contorno exige que, para a avaliação da força resultante sobre uma dada partícula, a interação com as demais partículas da caixa de simulação e a interação com as partículas das caixas imagem sejam incluidas. Consequentemente, o parâmetro referente às interações entre partículas será consti-tuído por infinitos termos, e, na prática, sua avaliação requer a realização de algumas aproximações [86, 90].
rc
FIGURA2.4: Caixa de simulação, sob condições periódicas de contorno com raio de corte,
rc.
Para o tratamento das interações de curto alcance, usualmente emprega-se um pro-cedimento denominado convenção da imagem mínima. Essa aproximação permite que apenas a interação com a imagem mais próxima seja incluída no cálculo das forças. Para tanto, é realizado o truncamento do potencial a partir de um raio de corte esférico, escolhido de tal forma que a partícula possa interagir somente com uma de suas ima-gens, e a interação com a mesma partícula duas vezes seja inviabilizada, como mostra a Figura 2.4. Para que isso ocorra, o raio de corte não pode ser maior do que a metade do menor comprimento da célula [86], ou seja,
Capítulo 2. Metodologia 14
Analogamente, para as interações de longo alcance, o truncamento abrupto do po-tencial produziria efeitos que, em muitos casos, comprometeriam severamente os resul-tados das simulações. Atualmente, este problema tem sido contornado com a utilização de métodos que produzem trajetórias estáveis [86].
2.1.7 Termostatização e barostatização
A aplicação direta da formulação desenvolvida na seção 2.1.2 simula um sistema iso-lado, ou seja, um ensemble NVE. Para simulação de outros ensembles no cálculo de propriedades, o algoritmo de integração deve ser modificado de tal forma que sejam incluídos os efeitos de controle de temperatura e de pressão. Esses controles são co-nhecidos como termostatização e barostatização, respectivamente.
Equações de movimento para o ensembleNVT
Um dos métodos para integração das equações de movimento mais amplamente di-fundido para se gerar uma distribuição canônica (NVT) é o de Nosé-Hoover. Essa me-todologia adiciona às equações do movimento algumas variáveis dinâmicas artificiais acerca de posição e de velocidade. Como resultado, obtém-se o controle da tempera-tura [91]. Embora possa ser demonstrado que este método gera um ensemble canônico adequado, em algumas circunstâncias, ele não tem se mostrado uma boa alternativa [92].
Nesses casos, é possível se adotar uma forma generalizada da abordagem de Nosé-Hoover [93]. Uma série de variáveis associadas ao banho térmico é adicionada ao es-paço de fases no intuito de se orientar as flutuações da energia cinética, de tal modo que, na média, se alcance o valor esperado.
As equações de movimento, a partir de então, podem ser reescritas como:
˙
rA= pA mA
(2.14)
˙
pA=FA−
pθ1
Q1
pA (2.15)
˙ θs=
pθs
Qs
s= 1,· · ·,ℵ (2.16)
˙ pθ1 =
" X
A
p2A ma −
3N kbT
#
−pθ1θ˙2 (2.17)
˙ pθs =
"
p2θ
s−1
Qs−1 − kBT
#
−pθsθ˙s+1 (2.18)
˙ pθℵ =
p2
θℵ−1
Qℵ−1 −
Capítulo 2. Metodologia 15
ondepθs é o momento associado aosℵtermostatos eQs uma inércia de cada
termos-tato scom dimensão de massa, eventualmente chamado de massa de Nosé. As Eq.
2.15 e 2.17 acoplam a evolução dinâmica dos termostatos ao movimento das partículas. Quanto maior o número de termostatos, maior o custo computacional.
2.1.8 Configuração inicial, minimização e equilibração
Uma vez que os sistemas fluidos não possuem arranjo atômico/molecular bem defi-nido como, por exemplo, num sistema sólido cristalino, a configuração inicial para as simulações pode ser estabelecida distribuindo-se aleatoriamente as partículas na caixa de simulação. Além das posições, as velocidades iniciais também devem ser definidas aleatoriamente levando-se em conta uma temperatura de referência. Por fim, os valo-res aleatórios das velocidades devem ser escalonados para que o sistema seja ajustado à temperatura especificada.
Um problema que geralmente ocorre numa geração aleatória de partículas é a so-breposição atômica ouoverlap. Esta sobreposição ou superproximidade entre algumas particulas resulta num valor muito alto para energia potencial do sistema no início da simulação. Em virtude de num sistema do tipo Lennard-Jones (seção 2.2.1) curtas dis-tâncias resultarem em forte repulsão, algumas partículas da caixa de simulação seriam projetadas para uma distância além dos limites das condições de contorno periódica. Se, nesse caso, o ensemble utilizado na simulação levasse em conta o número constante de partículas, a simulação já estaria invalidada.
Nesse contexto, é comum que os problemas de dinâmica molecular realizem no iní-cio da simulação uma etapa de minimização. Nessa etapa, a energia é minimizada pelo rearranjo iterativo dos átomos. O procedimento pode ser finalizado obedecendo algum critério de parada baseado, por exemplo, nas diferenças de energias ou nos módulos das forças das interações.
2.2
Potenciais de interação
O potenciais de interação ocupam papel central nas simulações de dinâmica molecular, já que, a partir deles, as propriedades do sistema podem ser calculadas. Por exemplo, muitas das propriedades dinâmicas dos materiais podem ser interpretadas em termos desses potenciais uma vez que dependem explicitamente das posições dos átomos. Dessa forma, a escolha do potencial de interação está intimamente relacionada com a qualidade dos resultados obtidos nos cálculos computacionais [94, 95]. É recomen-dável que os modelos de interação sejam os mais simples possível, a fim de se garantir que o número de parâmetros envolvidos seja pequeno [96]. Nesse contexto, destaca-se o potencial de Lennard-Jones que será discutido a seguir.
Capítulo 2. Metodologia 16
tipo caroço atenuado (Core-Softned potentials) tem permitido que as simulações repro-duzam, com sucesso, através de uma abordagem simples, o comportamento anômalo termodinâmico, dinâmico e estrutural. Esses potenciais serão abordados no decorrer do texto.
2.2.1 Potencial de Lennard-Jones (LJ)
Proposto em 1924 por John Lennard-Jones [97], o potencial considera apenas interações do tipo atração-repulsão e tem sido amplamente empregado para representar espécies moleculares apolares e simétricas. Para duas espécies distantes entre si de uma distân-ciar, o potencial LJ é definido como:
U(r) = 4ǫ
" σ
r
12
−
σ
r
6#
. (2.20)
O Potencial de Lennard-Jones é o modelo mais simples de potencial contínuo e pos-sibilita a determinação das forças de interação sem grandes dificuldades numéricas. Ele apresenta uma forte região repulsiva para distâncias pequenas, um valor mínimo numa zona intermediária e uma região atrativa com inclinação bastante suave para distâncias maiores. Para tanto, esse potencial apresenta apenas dois parâmetros,σ eǫ. O parâ-metroσestá intimamente relacionado com o diâmetro molecular e apresenta dimensão de comprimento. O parâmetroǫpor sua vez, tem dimensão de energia e representa a profundidade do poço de potencial associado à interação. O comportamento típico da energia potencial em função da distância descrito pela Equação 2.20 associado a esses parâmetros está apresentado na Figura 2.5.
σ
r
!
Capítulo 2. Metodologia 17
Algumas considerações importantes podem ser feitas com base na Figura 2.5 [98, 99]. Se duas partículas estão separados por distânciasr < r0, o potencial de interação U(r < r0) é puramente repulsivo, enquanto que, ser > r0 U(r > r0), trata-se de um potencial atrativo.r=r0determina a posição de equilíbrio estável da partícula. Nesse pontoU(r0), corresponde a um mínimo de energia associado a uma força nula, uma vez que dU(r0)
dr = 0. Ser→ ∞,U(r)é atrativo e decai comr6. Por outro lado, ser →0,
U(r) é repulsivo com r12. De fato, r12 diverge tão rapidamente que duas partículas raramente são capazes de se aproximar a distâncias inferiores aσ. Energias negativas muito próximas deU =ǫcorrespondem à pequenas oscilações em torno da posição de equilíbrio.
2.2.2 Potenciais do tipo caroço atenuado (Core-softened)
Os potenciais do tipo caroço atenuado (core-softened potentials) tem se tornado uma al-ternativa computacional mais barata para a simulação de fluidos anômalos, como a água, visto as limitações e os altos custos computacionais envolvendo modelos mole-culares clássicos e cálculos de primeiros princípios. Potenciais do tipo caroço atenuado tratam fluidos anômalos de forma eficaz através de uma abordagem mais simples. A água, por exemplo, tem o seu comportamento anômalo termodinâmico, dinâmico e estrutural muito bem reproduzido por esses modelos.
Cada molécula de água faz ligações de hidrogênio com quatro moléculas vizinhas, formando estruturas tetraédricas com distâncias e ângulos bem definidos denominadas tetrâmeros [100]. Os potenciais efetivos reduzem as duas distâncias preferenciais para os tetrâmeros de água e, dessa forma, cada partícula do fluido representa, de forma efe-tiva, um tetrâmero de água. Um outro ponto importante é que não há direcionalidade e nem a presença de cargas. Estes ingredientes transformam os modelos baseados nes-ses potenciais em ferramentas importantes e de baixo custo computacional. Esnes-ses tipos de modelos têm sido fortemente explorados não só computacionalmente mas também por meio de ferramentas teóricas devido à sua simplicidade[51–62].
Os potenciais de caroço duro atenuado possuem um caroço repulsivo, e uma mu-dança de curvatura no potencial introduz uma parte resulsiva atenuada, isto é, um ombro ou uma rampa [101–118]. A Figura 2.6(a) mostra um modelo tipo ombro for-mado por um caroço duro, um ombro repulsivo e um poço quadrado enquanto a Fi-gura 2.6(b) apresenta um potencial do tipo rampa repulsiva que define duas distâncias de equilíbrio competitivas.
Capítulo 2. Metodologia 18
r r
FIGURA2.6: Potenciais do tipo caroço atenuado descontínuos do tipo ombro (a) e do tipo
rampa(b).
modelo, de acordo com a Figura 2.7, duas escalas de interação se fazem presentes. O potencial surge da soma de uma gaussiana centrada emr0, de alturaae largurac, com o potencial de Lennard-Jones. O modelo foi definido para um sistema de partículas idênticas, de diâmetroσ, de tal forma que
U∗(r) = U(r) ǫ = 4
" σ
r 12
−
σ r
6#
+aexp "
−c12
r
−r0
σ 2#
. (2.21)
0 1 2 3 4 5 6
0 1 2 3 4 5 6
r
U
∗(r
)
FIGURA2.7: Potencial isotrópico de ombro repulsivo desenvolvido por de Oliveira e cola-boradores e descrito pela Equação 2.21. Os parâmetros utilizados forama= 5, r0/σ= 0.7
ec= 1.
Capítulo 2. Metodologia 19
Os sucessivos bons resultados deram suporte ao uso de potenciais do tipo caroço atenuado em ambientes mais complexos, como, por exemplo, no confinamento de água entre placas [63–65].
Miha Luksic e colaboradores reproduziram computacionalmente uma solução sa-lina e estudaram como a presença de íons pode alterar o conjunto de anomalias es-truturais, termodinâmicas e dinâmicas de um fluido. O solvente foi simulado através de potenciais do tipo caroço atenuado, enquanto os íons positivos e negativos foram modelados como esferas rígidas carregadas [66]. O estudo permitiu, dentre outras con-clusões, a determinação das regiões que delimitam as anomalias estruturais e termodi-nâmicas para esse modelo de mistura.
Capítulo 3
O Modelo
Neste capítulo são apresentados os detalhes a respeito do modelo efetivo de separação soluto solvente proposto e também as principais quantidades de interesse que serão analisadas.
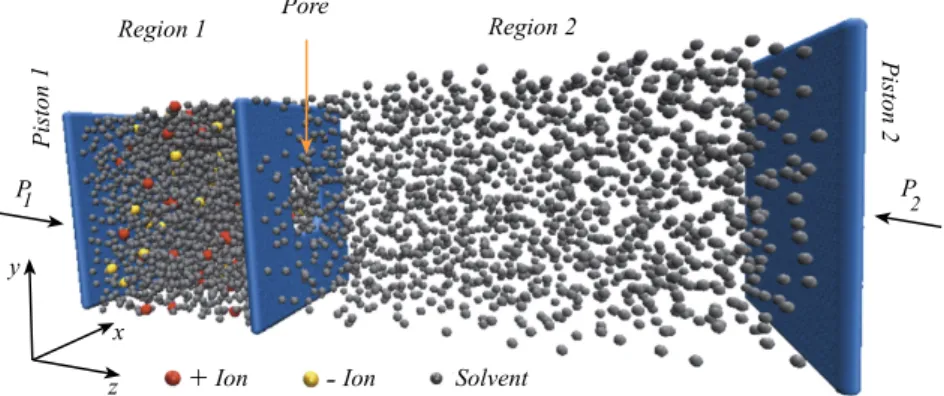
O sistema construído para simular o processo de separação soluto-solvente é apre-sentado pela Figura 3.1. Foi utilizada uma caixa virtual de simulação com dimensões de 25×25×75, em unidades deσ (tamanho de partículas de solvente), nas direçõesx, yez,respectivamente, e dois pistões, um localizado inicialmente em z = 0 (pistão 1) e outro em z = 75σ(pistão 2). Em todas as simulações pressões externas foram aplicadas no pistão 1. O pistão 2, por sua vez, pode ser estático ou sofrer pressões externas, como será discutido a seguir. Finalmente, há uma parede do meio cuja posição é fixa em z = 25σ (membrana), e que separa a Região 1 (reservatório de alimentação) da Região 2 (permeado). Condições de contorno periódicas foram utilizados na direçõesxey. Os pistões e a parede foram constituídas por 625 partículas "congeladas", igualmente dis-tribuídas em uma única camada. O termo "congelados"refere-se ao fato de que a força resultante que age sobre essas partículas é igual a zero. No caso específico da parede do meio, algumas das suas partículas foram eliminadas em um certo estágio das simu-lações para construção de um poro central no intuito de que as partículas de solvente pudessem passar para a Região 2. O número de partículas eliminadas depende da área do poro em estudo.
Inicialmente 3000 partículas de solvente e 120 partículas de soluto (60 partículas carregadas positivamente e 60 partículas carregadas negativamente) foram colocadas aleatoriamente na Região 1, enquanto que a Região 2 foi inicialmente mantida vazia ou contendo 1500 partículas de solvente, dependendo do problema (isto será discutido em detalhes à frente). Como resultado das pressões externas aplicadas no pistão 1, as partículas eventualmente atravessaram o poro circular. De modo a evitar flutuações de pressão excessivas no fluido, as partículas que constituem os pistões e a membrana foram modeladas com massa igual à 1000 vezes a massa das partículas de solvente. Além disso, uma força viscosa proporcional à velocidade dos pistões foi usada com constante de proporcionalidade 100 em unidades reduzidas. As partículas de soluto e solvente foram modeladas tendo a mesma massa.
Capítulo 3. O Modelo 21
Poro
Região 1
Região 2
Ion -
Solvente
P
is
tão 1
x
y
z
1
P
P
is
tão 2
P
2
Ion +
FIGURA3.1: Sistema construído para simular o processo de separação soluto-solvente.
Energias e comprimentos são dados em termos dos parâmetros de Lennard-Jones: ǫ(energia) eσ(comprimento), respectivamente. Todas as outras quantidades investi-gadas podem ser reduzidas para formas adimensionais escritas em termos deǫ,σ, m e constante de BoltzmannkB. Por exemplo, a pressãoP é dada em termos deǫ/σ3, o
tempoτ em termos dep
mσ2/ǫ, e a temperatura em termos deǫ/k
B.
Simulações de dinâmica molecular foram realizadas com o software livre de código aberto LAMMPS(Large-scale Atomic/Molecular Massively Parallel Simulator)[125], com o ensemble estatístico canônico (NVT) gerado pelo método de termostatização de Nosé-Hoover [126]. Foram estudados dois casos diferentes: (i) caso 1, em que o pistão 2 é mantido imóvel, enquanto o pistão 1 comprime o fluido e (ii) caso 2, onde são aplica-das pressões externas em ambos os pistões. Em especial para o caso 1, ora a região 2 inicialmente estava vazia, ora apresentava 1500 partículas de solvente.
As simulações foram divididas em estágios de equilíbrio e de produção. Em ambos os casos, o estágio de equilíbrio foi subdividido em dois subestágios como se segue. Primeiramente, o sistema foi simulado durante 150.000 passos com temperatura inicial T = 2 e temperatura final T = 1 (o poro foi mantido bloqueado durante toda a fase de equilibração, afim de evitar que partículas de solvente atingissem a região 2). Depois disso, o sistema foi simulado por mais 150.000 passos em T = 1, e a energia de configu-ração e a pressão mostraram ser estáveis, ou seja, flutuar em torno de valores médios. Além disso, no caso 2, o sistema foi simulado por mais de 200.000 passos em T = 1 com uma pressão externa aplicada ao Pistão 2. Esta pressão foi escolhida de modo a igualar a pressão osmótica do fluido a T = 1 e a uma densidade referente a 1.500 partículas confinadas na Região 2.
Capítulo 3. O Modelo 22
sistema até5×106passos, dependendo da quantidade de interesse em investigação. O passo de tempo utilizado em todas as execuções foi de 0,001 em unidades reduzidas. Todas as propriedades de interesse foram calculadas sobre a média de cinco simulações independentes e as barras de erro calculadas para as quantidades de interesse foram menores do que símbolos para todas as figuras.
As interações entre os constituintes do sistema foram definidas como se segue. A interação entre as moléculas de solvente foi dada pelo o potencial efetivo (descrito na seção 2.2.2) proposto por de Oliveira e colaboradores [119, 120]:
Uss∗(rss) =
Uss(rss)
ǫ = 4
" σ rss 12 − σ rss 6#
+aexp "
−c12
r
−r0 σ
2#
. (3.1)
σ e ǫ são parâmetros de Lennard-Jones enquanto a = 5, r0/σ = 0.7 e c = 1 estão relacionados com a forma do termo gaussiano. rss é a distância entre as partículas de solvente.
A interação soluto-soluto foi descrita pela parte repulsiva do potencial de Lennard-Jones mais forças de Coulomb. Ele pode ser descrito pela equação
Uii ǫ′ =C
z1z2 rii
+ Θ(σ′−rii)uLJ(rii), (3.2)
ondeΘ(R)é a função de Heaviside (ou função degrau), em queΘ(R <0) = 0,Θ(R≥ 0) = 1, e
uLJ(rii) = 4
" σ′ rii 12 − σ′ rii 6# . (3.3)
z1 ez2 receberam os valores+1e−1, de acordo com a valência dos íons. σ′ = 3σ e ǫ′ =ǫ. C/σ′ = 1/4é uma constante associado à força do termo de Coulomb. O raio de corte para o potencial de Coulomb foi de 7.5σ′ [127]. As interações foram calculadas diretamente entre pares de partículas para distâncias menores que o raio de corte. Para distâncias maiores que o ponto de corte, foi utilizado o esquemaParticle Particle-Particle Mesh[128]. A interação entre as partículas de solvente e de soluto foi descrita por um potencial de Lennard-Jones,
Usi ǫ′′ = 4
" σ′′ rsi 12 − σ′′ rsi 6# (3.4)
comσ′′ = (σ+σ′)/2 = 2σ. O parâmetroǫ′′foi baseado em um trabalho desenvolvido por Luksic e colaboradores [66] em que as propriedades estruturais e termodinâmicos de um modelo de solvente-soluto muito semelhante foram investigadas por meio de Monte Carlo e Equações Integrais. Neste trabalho, a interação solvente-soluto foi de-finida porǫ′′/k
BT = (Ts∗Tel∗)−1/2. Ts∗ = kBT /ǫé a temperatura reduzida do solvente,
T∗
Capítulo 3. O Modelo 23
e λB = Cǫ/kBT é o comprimento de Bjerrum. Depois de alguns cálculos, obtem-se
ǫ′′=ǫp
C/σ′ =ǫ/2. Este modelo é composto por esferas duras carregadas com cargas positivas e negativas. O solvente, geralmente água, é representado por uma constante dielétrica e muda a permissividade elétrica do meio. A interação de Coulomb entre os íons leva em conta, ao invés deǫ0, umǫdependente do solvente, da pressão e da tem-peratura. O modelo restrito primitivo considera o diâmetro dos íons positivos igual ao diâmetro dos íons negativos.
As partículas que compõem os pistões e a parede interagem com as partículas de solvente e soluto através da parte repulsiva do potencial de Lennard-Jones. O tamanho das partículas que compõe os pistões e a parede foi definido como 2σ.
O sistema com o pistão 2 imóvel (caso 1) também foi estudado sob efeito de um campo elétrico externo aplicado paralelamente aos pistões. Esse processo, conhecido como deionização capacitiva, é tido hoje como uma alternativa de alta eficiência no que se refere ao processo de dessalinização de água [129, 130] A deionização capaci-tiva baseia-se na remoção dos íons presentes em solução armazenando-os na interface eletrodo-solução através da aplicação de uma diferença de potencial [25]. Este processo pode ser observado na Figura 4.1.1.
água
FIGURA3.2: Esquema representativo do processo de deionização capacitiva. Uma solução
contento íons atravessa dois eletrodos submetidos a uma diferença de potencial. Os cátions são atraídos para o eletrodo negativo e os ânions são atraídos para o eletrodo positivo.
Um dos pontos que tem sido explorados na literatura é a possibilidade dos poros de membranas de grafeno serem passivados com hidrogênios, grupos hidroxila, nitrogê-nio e outras substâncias [20, 23, 131]. No caso do modelo desenvolvido neste trabalho, esta funcionalização foi representada pela alteração das interações entre fluido e a fron-teira do poro (primeira camada de partículas), como pode ser observado na Figura 3.3. A interação entre essas partículas e as partículas de soluto e solvente foi represen-tada por um potencial de Buckingham, descrito pelo equação
Usb=Ae−r/ρ− C rsb6
(3.5)
Capítulo 3. O Modelo 24
FIGURA3.3: Construção da membrana com poro central com a primeira camada de áto-mos (borda) diferenciada. A alteração do potencial de interação da borda representa a funcionalização ou a passivação do poro.
partículas da borda e as partículas de soluto os parâmetros foram definidos comoA= ρ=C = 0, já para as partículas de solventeA=−1,ρ= 0,5eC= 0.
A Figura 3.4 apresenta o comportamento da energia potencial de interação em fun-ção da distância para o potencial de Buckingham descrito pela Equafun-ção 3.5 e pelos parâmetros indicados anteriormente para a interação da borda com as partículas de solvente.
1 1.5 2 2.5 3
−0.4
−0.3
−0.2
−0.1 0
r
U
(
r
)
FIGURA 3.4: Comportamento da energia potencial de interação em função da distância
para o potencial de Buckingham descrito pela Equação 3.5 e pelos parâmetros indicados anteriormente para a interação da borda com as partículas de solvente,A=−1,ρ= 0,5e
C= 0.
3.1
Quantidades de interesse
Capítulo 3. O Modelo 25
a membrana como uma função de tempo, e (iii) a dependência fluxo volumétrico de solvente com a pressão externa aplicada.
A rejeição de soluto da membrana foi calculada conforme a equação:
R=
1−N1/2 N0
(3.6)
onde N0 é o número inicial de partículas de soluto na Região 1 e N1/2 é o número de partículas de soluto na região 2 no tempo em que metade das partículas de solvente passaram da Região 1 para a Região 2. Esta definição coincide com a utilizada por Chen e colaboradores [23] and Zhu e colegas [21] em trabalhos que utilizaram simulações de dinâmica molecular para estudar o comportamento da água e dos íons passando através de nanomembranas. De acordo com a Equação 3.6 , seN1/2 = 0 a membrana apresenta 100 % de rejeição de soluto ouR = 1. Por outro lado, para uma membrana que não apresenta rejeição de soluto,R= 0, o número de partículas de soluto na Região 2 deve ser idênticas aN0.
O fluxo volumétrico de solvente (φ) é o volume de fluido por unidade de tempo que passou para a Região 2 (dV2/dt) e dá uma estimativa da eficiência da configuração do sistema. Ele pode ser calculado através da equação
φ= 1 ρ1
dN
dt (3.7)