*Correspondence: A. M. de Souza. Laboratório de Hematologia. Departamento de Análises Clínicas, Toxicológicas e Bromatológicas. Faculdade de Ciências Farmacêuticas de Ribeirão Preto. Universidade de São Paulo. Av. do Café, s/n - Monte Alegre - Bloco M, 20 andar, Sala 053-A. 14040-903 - Ribeirão Preto, SP, Brasil. E-mail: [email protected]
A
vol. 51, n. 2, apr./jun., 2015 http://dx.doi.org/10.1590/S1984-82502015000200013
Prevalence of hemoglobinopathies in school children: the
importance of using confirmatory methods
Cristiane Fernandes de Freitas Tavares, Jacqueline da Silva Guimarães, Ana Maria de Souza
*Department of Clinical Analysis, Toxicology and Food Science, Faculty of Pharmaceutical Sciences of Ribeirão Preto, University of São Paulo, Ribeirão Preto, SP, São Paulo, Brazil
The hemoglobinopathies are included among the most common genetic diseases in the world. In Brazil, hemoglobinopathies are related to the diversity of racial backgrounds and the degree of interbreeding. The study focused on the prevalence of hemoglobinopathies using conventional and conirmatory laboratory tests in children from public schools in Ribeirão Preto-SP. The study involved the participation of 427 children between six and nine years of age. Hematologic evaluation, hemoglobin electrophoresis on cellulose acetate at alkaline pH, quantiication of hemoglobin fractions by high performance liquid chromatography (HPLC) and detection of -α3.7 deletion for α thalassemia by polymerase chain reaction
were performed. The results of hemoglobin electrophoresis on cellulose acetate and HPLC of the children studied showed the presence of 30 children (7%) with hemoglobinopathies. Eleven children presented results indicating suspicion of S/β-thalassemia; their parents and/or siblings were evaluated and conirmed the presence of only Hb S. The analysis of deletion -α3.7 to characterize α-thalassemias
sampling performed on 207 participants identiied 26 children (12.6%) with deletion -α3.7. Thus, 54
(12.6%) of the children studied present this genetic alteration. For the detection of α-thalassemias it is necessary to use conirmatory methods such as molecular analysis and evaluation of family members in doubtful cases to facilitate genetic counseling in families, in which deletion -α3.7 is more frequent in Brazil.
Uniterms: Hemoglobinopathy. Hemoglobin electrophoresis. High performance liquid chromatography/ quantitative analysis. Polymerase chain reaction.
As hemoglobinopatias estão incluídas nas doenças genéticas mais comuns no mundo. No Brasil, as hemoglobinopatias são relatadas pela diversidade racial e o grau de miscigenação. O estudo focou a prevalência das hemoglobinopatias usando métodos laboratoriais convencionais como a eletroforese de hemoglobina em acetato de celulose em pH alcalino e conirmatório por reação em cadeia de polimerase (PCR) em crianças de escolas públicas de Ribeirão Preto-SP. O estudo envolveu a participação de 427 crianças entre 6-9 anos de idade. Determinaram-se os valores hematológicos, efetuou-se eletroforese de hemoglobina em acetato de celulose em pH alcalino, quantiicação das frações de hemoglobina por HPLC e a detecção da deleção -α3,7 pela PCR. Os resultados da eletroforese de hemoglobina em
acetato de celulose e do HPLC, nas crianças estudadas, mostraram a presença de 30 crianças (7%) com hemoglobinopatias. Onze crianças apresentaram resultado indicando a suspeita de S/β-talassemia; seus
pais e/ou irmãos conirmaram a presença de apenas a Hb S. A análise da deleção -α3,7, uma das alterações
que estão presentes na α-talassemia, realizada em 207 participantes, identiicou 26 crianças (12,6%) com a deleção -α3,7. Dessa forma, 54 (12,6%) das crianças estudadas apresentam hemoglobinopatias. Para
a deleção da α-talassemias é necessário utilizar métodos conirmatórios como as análises moleculares e avaliação de membros da família, em casos duvidosos, facilitando o aconselhamento genético nas famílias, sendo a deleção -α3,7 mais frequente no Brasil.
Unitermos: Hemoglobinopatias. Eletroforese de hemoglobina. Cromatograia líquida de alta eiciência/
INTRODUCTION
The hemoglobinopathies are a group of inherited diseases that are classified based on the presence of structurally abnormal hemoglobin (Hb) such as hemoglobins S, C, D and E, and/or one or more globin chain disabilities, known as thalassemias (Clark, Thein,
2004; Henderson et al., 2009). These pathologies are included among the most common genetic diseases in the world, with an estimated prevalence of 7% of the worldwide population (Melo-Reis et al., 2006; Manca,
Masala, 2008).
In Brazil, hemoglobinopathies are related to the diversity of racial backgrounds and the degree of interbreeding, and may be regional. The most frequent hemoglobin variants are S and C of African origin, however, due to interbreeding these hemoglobins came to be found in other ethnic groups (Aigneret al., 2006). The
impact of population migration increases the combinations of abnormal hemoglobins and the variety of mutations (Henderson et al., 2009).
In most abnormal hemoglobins a point mutation, in other words, a single amino acid substitution occurs.
Over 1100 hemoglobin variants involving the chains α, β, δ and γ have been described (Wagner et al., 2005). In the heterozygous state, production of Hb A and variant hemoglobin occurs without severe clinical manifestations. In the homozygous state, Hb A is absent, thus possibly resulting severe anemia. The interaction of two hemoglobin variants or a combination of a hemoglobin variant and a genetic alteration to thalassemia may still occur (Clark, Thein, 2004).
Different types of thalassemia are found in
the population, the most common being the α and β-thalassemias (Galanello et al., 1998; Galanello, Origa, 2010; Cousenset al., 2010). The α-thalassemia is the most
common inherited disease in the world. Recent studies
show that α-thalassemia affects 5% of the worldwide
population and until recently (Vichinsky, 2010), the frequency of this pathology was underestimated due the use
of inadequate diagnostic methods. In Brazil α-thalassemia
affects mainly Asian and some African groups. Studies show a frequency of 10-12% in some regions and up to
25% in speciic groups. For α-thalassemia in the state of
São Paulo in Caucasian descendants, the estimated ratio is 3% (Bonini-Domingos, 2004).
Mutations or deletions may result in a lack of
production of globin (β0 and α0) or decreased production
of these (β+ and α+) (Cunninghan, 2010). Over 95% of
cases of α-thalassemia are due to deletions, being -α5.2
and -α 20.5 generally causing α0-thalassemia and -α3.7, -α4.2
causing α+-talassemia (Weatherall, 2001). Molecular
analyses indicate a high prevalence of deletion -α3.7 in the
state of São Paulo (Sonati et al., 1991; Borges et al., 2001;
Oliveira et al., 2006).
Conventional laboratory diagnosis for α-thalassemia
can be characterized by precipitation of hemoglobin H, visualized by supravital staining. This diagnosis can be
dificult when there is a concomitant hemoglobinopathy
such as Hb S, Hb C, Hb E and β-thalassemia, because of the decrease or absence of Hb H and cellular inclusions (Chui et al., 2003).
In general, the most used method for the identiication
of hemoglobinopathies is hemoglobin electrophoresis on cellulose acetate at alkaline pH, however its sensitivity is limited, especially in the case of isoform co-migration, such as Hb S, Lepore and Hb D (Chinelato-Fernandes
et al., 2003). Thus, conirmatory tests are required, such
as acid agarose gel electrophoresis, although there is controversy regarding the distinction of hemoglobins D
and G, and O and E (Ondei et al., 2007). High-performance liquid chromatography (HPLC) and molecular techniques such as polymerase chain reaction (PCR) and sequencing the gene are more accurate methods for the diagnosis of
hemoglobinopathies (Bertholo, Moreira, 2006; Wenning et al., 2000; Bonini-Domingos, 2004).
The combined analysis of all methods in the diagnosis of hemoglobinopathies is the most appropriate since each method alone has limitations and can cause a misinterpretation of results (Hughes et al., 2009). Thus, this study aimed to evaluate the prevalence of
hemoglobinopathies using conventional and conirmatory
laboratory tests such as hemoglobin electrophoresis, quantification of hemoglobin fractions by HPLC, and analysis of the deletion that causes more frequent
α-thalassemia (-α3.7) in southeast Brazil (Wenning et al.,
2000; Bonini-Domingos, 2004), also occurring in children
from public schools in Ribeirão Preto-SP, by molecular biology.
MATERIAL AND METHODS
The study group included 427 children between six and nine years of age of both genders, recruited from state and municipal schools in the city of Ribeirão Preto-SP, namely: Centro Municipal de Educação Infantil Virgílio Salata, Escola Estadual Antônio Diederichsen,
Escola Estadual Dr. Tomas Alberto Whatelly, EMEFEM
Dom Luiz Do Amaral Mousinho, and Escola Estadual
Dom Alberto José Gonçalves. Participation in this study
Dentistry of Ribeirão Preto, University of São Paulo (case
no. 2006.1.797.58.5).
Ten milliliters (mL) of blood were collected a n d p l a c e d i n a t u b e c o n t a i n i n g d i p o t a s s i u m ethylenediaminetetraacetic acid (K2 EDTA) 10%, which was used in the following determinations: hematologic evaluation using automatic counter Micros 45- Horiba ABX®; hemoglobin electrophoresis on cellulose acetate
at alkaline pH, according to Naoum (1999); quantiication
of hemoglobin fractions by HPLC Variant II Bio-Rad®
automated system (β-thalassemia Short Program kit); and
extraction of genetic material to perform the PCR.
For detection of deletion -α3.7 the speciic primers
(Dodé et al., 1993): 5’ CCA TGC CTG GCA CGT TTG CTG AGG 3’(C9 primer); 5’ GAT GCA CCC ACT GGA
CTC CT 3’(C10 primer) were used. The PCR reaction was processed in a thermocycler (Eppendorf Mastercycle), 30 cycles being performed at 94 oC/2 min., 56 oC/1 min., 72 oC/2 min., preceded by an initial heat-denaturation step
at 94 oC/5 min. and followed by a inal extension step at
72 oC/10 min. The mixture was subjected to horizontal
electrophoresis on a 1% agarose gel with ethidium bromide, with a running time of 1 hour and 20 minutes at 90 volts, 220 mA. Then the gel was exposed to UV light for visualization of the presented bands.
Thirteen parents and two brothers participated in the study in order to confirm the results of children who showed the presence of Hb S and increased Hb A2,
suggestive of hemoglobinopathy HbS/β-thalassemia.
Five mL of blood were collected and placed into a tube containing K2 EDTA, which was used for the identiication
and quantiication of hemoglobin fractions by HPLC.
RESULTS AND DISCUSSION
The results of hemoglobin electrophoresis on cellulose acetate and HPLC of the 427 children studied indicated the presence of 30 children (7%) with hemoglobinopathies, being 14 with increased Hb A2,
consistent with the phenotype of β-thalassemia (β+ or
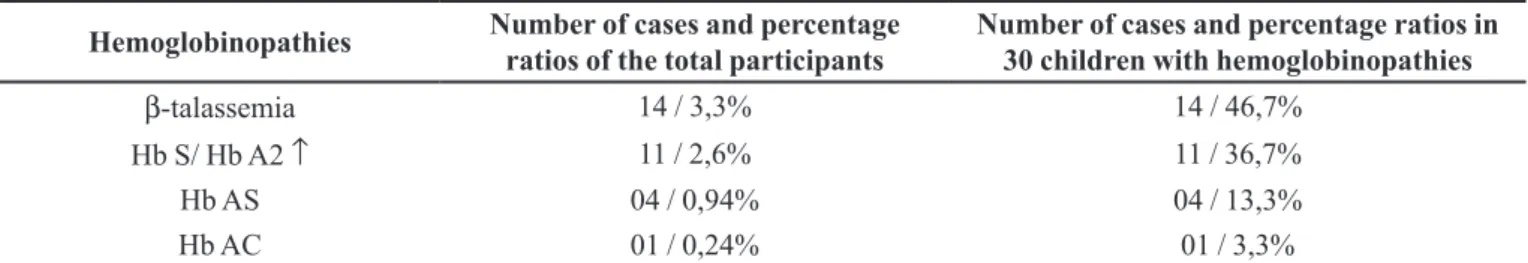
TABLE I - Distribution of hemoglobinopathies in the studied children utilizing hemoglobin electrophoresis on cellulose acetate and HPLC methodology
Hemoglobinopathies Number of cases and percentage
ratios of the total participants
Number of cases and percentage ratios in 30 children with hemoglobinopathies
β-talassemia 14 / 3,3% 14 / 46,7%
Hb S/ Hb A2 ↑ 11 / 2,6% 11 / 36,7%
Hb AS 04 / 0,94% 04 / 13,3%
Hb AC 01 / 0,24% 01 / 3,3%
FIGURE 1 - Distribution of hemoglobinopathies in the 427 studied children utilizing hemoglobin electrophoresis on cellulose acetate and HPLC methodology.
β0); 11 with increased Hb A2 and the presence of Hb S,
consistent with the phenotype of Hb S/β-thalassemia; 4
with the presence of Hb S carriers of sickle cell trait and 1 with the presence of Hb C, indicating heterozygosity for the Hb C disease (Table I, Figure 1).
The determination of the levels of Hb A2 of 11
participants with suspected Hb S/β-thalassemia was
performed by HPLC and the fractions obtained by elution in electrophoresis on cellulose acetate. The median Hb
A2 levels determined by HPLC and elution were 6.3% (± 1.1) and 7.0% (± 1.6), respectively, with no signiicant
statistical difference (p > 0.05). A survey of quantitative and qualitative changes of hemoglobin fractions in 11 parents and/or siblings by HPLC only detected the
presence of Hb S, discarding the suspicion S/β-thalassemia
in children.
The analysis of deletion -α3.7, featuring α-thalassemia
was performed in 207 study participants verifying the
presence of 26 children (12.6%) carrying the deletion,
being 24 heterozygous and 2 homozygous.
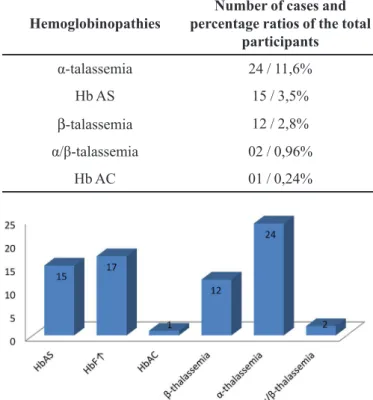
Together the analyses showed a total of 54 children,
12.6% of the children studied, showing some qualitative
or quantitative change of the globin chains, namely: 15 (3.5%) women with Hb S, 1 (0.24 %) Hb C and 14 (3.3%)
Among patients with hemoglobinopathies, 27
(50%) were anemic according to WHO criteria, i.e., Hb
less than 11.5 g/dL for children under 12 years of age of both genders, 8 (14.8%) of them showed only mild hypochromia and the remainder (35.2%) showed no
signiicant changes.
In our study the number of carriers of the sickle cell trait (3.5%) was apparently higher than reported in a review by Murao and Ferraz (2007), in which the authors estimate 1.9% of patients with Hb S in the state of Sao Paulo. A prevalence of approximately 3% of patients
with β-thalassemia in the state of São Paulo is estimated
(Bonini-Domingos, 2004). In our study a prevalence of 3.3%, very close to the above, was observed. There was
the presence of 12.6% of α-thalassemia, results close to the
literature (Head et al., 2004), which indicates prevalence of between 10 and 20% in our population. These numbers
are quite signiicant and justify the need for more extensive
research in the city of Ribeirão Preto to characterize the prevalence in the population.
On these results the necessity of conducting prenatal
testing and research of family cases of hemoglobinopathies in our population is evident.
Neonatal screening programs follow the standards of the National Program for Newborn Screening (Ministry of Health decree No. 822/01) that recommends the use of two presumptive methods for determining the electrophoretic profile. Currently, screening programs replaced the traditional methods by using isoelectric focusing electrophoresis and HPLC. Despite new presumptive methods having better sensitivity and
speciicity, they are susceptible to interference and in some cases such as in the diagnosis of α-talassemia the use of
molecular methodology is required (Hughes et al., 2009). Our study proved this since the number of patients with
hemoglobinopathies rose 5.6% after performing PCR for detection of α-thalassemia.
The use of HPLC has shown to be more sensitive and specific, and with a better reproducibility for the determination of the Hb A2 method. However it is argued that the percentage of Hb A2 measured by HPLC can be affected by the presence of hemoglobin S (HbS) in heterozygous individuals for β-thalassemia. This is due
to reduced afinity of the βS to α chains, which leads to
the free α to match δ chains, increasing Hb A2, and
co-elution of Hb with Hb A2, falsely elevating its percentage (Head et al., 2004; Kalleas et al., 2007). No signiicant
differences were observed in quantifying the levels of Hb A2, performed by the methods of electrophoresis
on cellulose acetate for elution and HPLC. To conirm suggestive cases of Hb S/β-thalassemia, parents and/or
siblings of these children were evaluated, verifying in all cases only the presence of HbS and normal levels of Hb A2, thus inferring that the children had only the sickle cell trait. Probably the increase in Hb A2 was found in these
children due to the combination of free α and δ chains and
not presenting mutations that compromise the synthesis
of β chains.
CONCLUSION
We c o n c l u d e t h a t i n o r d e r t o d e t e c t
hemoglobinopathies, the use of confirmatory methods
such as molecular analysis for the search of α-thalassemia
and the investigation of relatives of the doubtful cases are required. Although it has been investigated only for the
most frequent deletion for α-thalassemia, α3.7 it is now
possible to state the importance of conirmatory methods.
Only then we can ensure accurate diagnosis and facilitate genetic counseling in families where there are carriers of these pathologies.
TABLE II - Distribution of hemoglobinopathies in the studied children utilizing hemoglobin electrophoresis on cellulose acetate, HPLC and PCR methodology
Hemoglobinopathies
Number of cases and percentage ratios of the total
participants
α-talassemia 24 / 11,6%
Hb AS 15 / 3,5%
β-talassemia 12 / 2,8%
α/β-talassemia 02 / 0,96%
Hb AC 01 / 0,24%
ACKNOWLEDGMENT
The authors wish to thank CNPq for financial support.
REFERENCES
AIGNER, C.P. ; SANDRINI, F.; DUARTE, E.G.; ANDRADE,
M.P. ; LARGURA, M. A.; LARGURA, A. Estudo do peril
de hemoglobinas em 9.189 testes realizados no Álvaro Centro de Análises e Pesquisas Clínicas. Rev. Bras. Anal. Clin., v.38, n.2, p.107-109, 2006.
BERTHOLO, L.C.; MOREIRA, H.W. Focalização isoelétrica na
identiicação das hemoglobinas. J. Bras. Patol. Med. Lab.,
v.42, n.3, p.163-168, 2006.
BONINI-DOMINGOS, C.R. Thalassemia screening in Brazil:
results for 20 years. Rev. Bras. Hematol. Hemoter., v.26, n.4, p.288-289, 2004.
BORGES, E.; WENNING, M.R.; KIMURA, E.M.; GERVASIO, S.A.; COSTA, F.F.; SONATI, M.F. High prevalence of
alpha-thalassemia among individuals with microcytosis and hypochromia without anemia. Braz. J. Med. Biol. Res.,
v.34, n.6, p.759-762, 2001.
CLARK, B.E.; THEIN, S.L. Molecular diagnosis of haemoglobin
disorders. Clin. Lab. Haematol., v.26, n.3, p.159-176, 2004.
CHINELATO-FERNANDES, A.R.; LEONELI, G.G.; CALDERAN, p.O.; OLIVEIRA, R.B.; SILVA, W.A.; HIDALGO, C.A.; BONINI-DOMINGOS, C.R. Avaliação eletroforética, cromatográica e molecular da Hb D Los
Angeles no Brasil. Rev. Bras. Hematol. Hemoter., v.25, n.3,
p.161-168, 2003.
CHUI, D.H.K.; FUCHAROEN, S.; CHAN, v.Hemoglobin H
disease: not necessarily a benign disorder. Blood, v.101, n.3, p.791-799, 2003.
COUSENS, N.E.; GAFF, C.L.; METCALFE, S.A.;
DELATYCKI, M.B. Carrier screening for beta-thalassaemia: a review of international practice. Eur. J. Hum. Gen., v.18, n.10, p.1077-1083, 2010.
CUNNINGHAN, M.J. Update on thalassemia: clinical care and
complications. Hematol. Oncol. Clin. North Am., v.24, n.1, p.215-227, 2010.
DODÉ, C.; KRISHNAMOORTHY, R.; LAMB, J.; ROCHETTE,
J. Rapid analysis of –α3.7 thalassaemia and ααα anti 3.7
triplication by enzymatic amplification analysis. Br. J. Haematol., v.83, n.1, p.105-111, 1993.
GALANELLO, R.; SOLLAINO, C.; PAGLIETTI, E.; BARELLA, S.; PERRA, C.; DONEDDU, I.; PIRRONI,
M. G.; MACCIONI, L.; CAO, A. α-Thalassemia carrier
identification by DNA analysis in the screening for thalassemia. Am. J. Hematol., v.59, n.4, p.273-278, 1998.
HEAD, C. E.; CONROY, M.; JARVIS, M.; PHELAN, L.;
BAIN, B. J. Some observations on the measurement of haemoglobin A2 and S percentages by high performance liquid chromatography in the presence and absence of
α-thalassemia. J. Clin. Pathol., v.57, n.3, p.276-280, 2004.
GALANELLO, R.; ORIGA, R. Beta-thalassemia. Orphanet. J.
Rare Dis., v.5, n.11, p.2-15, 2010.
HENDERSON, S.; TIMBS, A.; MCCARTHY, J.; GALLIENNE, A.; MOURIK, M.V. ; MASTERS, G.; MAY, A.;
KHALIL, M.S.; SCHUCH, A.; OLD, J. Incidence of
haemoglobinopathies in various populations – the impact of immigration. Clin. Biochem., v .42, n.18, p.1745-1756, 2009.
HUGHES, H.Y.; MCKIE, K.; CARMICHAEL, H.; BORA, K.; KUTLAR, A.; KUTLAR, F. Diagnostic complication
and molecular characteristics of Hb SC-Chicago disease
with α-thal-2 (α3.7 deletion): effects of multiple variant on
patient’s phenotype. Ann. Hematol., v.88, n.11, p.1151-1153, 2009.
KALLEAS, C.; TENTES, I.; MARGARITIS, D.; et al. Effect of
HbS in the determination of HbA2 with the Biorad Variant II analyzer. Clin. Biochem., v.40, n.9-10, p.744-746, 2007.
MANCA, L.; MASALA, B. Disorders of the synthesis of human
fetal hemoglobin. IUBMB Life, v.60, n.2, p.94-111, 2008.
MELO, L.M.S.; SIQUEIRA, F.A.M.; CONTE, A.C.F.; BONINI-DOMINGOS, C.R. Rastreamento de hemoglobinas
MELO-REIS, p.R.; NAOUM, p.C.; DINIZ-FILHO, J.A.F.; DIAS-PENNA, K.G.B.; MESQUITA, M.M.; BALESTRA,
F.A. Prevalência de talassemias e hemoglobinas variantes
no estado de Goiás, Brasil. J. Bras. Patol. Med. Lab., v.42,
n.6, p.425-430, 2006.
MURAO, M.; FERRAZ, M.H.C. Sickle cell trait: heterozygous
for the hemoglobin S. Rev. Bras. Hematol. Hemoter., v.29, n.3, p.223-225, 2007.
NAOUM, p.C. Eletroforese: técnicas e diagnósticos. In: NAOUM, p.C. Hemoglobinopatias. 2.ed. São Paulo: Livraria Editora Santos, 1999. p. 57-100.
OLIVEIRA, G.L.V. ; MENDIBURU, C.F.; BONINI-DOMINGOS, C.R. Avaliação do peril hematológico de portadores de talassemia alfa provenientes das regiões
Sudeste e Nordeste do Brasil. Rev. Bras. Hematol. Hemoter.,
v.28, n.2, p.105-109, 2006.
ONDEI, L.S.; ZAMARO, p.J.A.; MANGONARO, p.H.; VALÊNCIO, C.R.; BONINI-DOMINGOS, C.R. HPLC
determination of hemoglobins to establish reference values with the aid of statistics and informatics. Genet. Mol. Res.,
v.6, n.2, p.453-460, 2007.
SONATI, M.F.; FARAH, S.B.; RAMALHO, A.S.; COSTA, F.F.
High prevalence of alpha-thalassemia in a black population of Brazil. Hemoglobin, v.15, n.4, p.309-311, 1991.
VICHINSKY, E. Complexity of alpha thalassemia: growing health problem with new approaches to screening, diagnosis, and therapy. Ann. NY Acad. Sci., v.1202, v.180-187, 2010.
WAGNER, S.C.; SILVESTRI, M.C.; BITTAR, C.M.; FRIEDRISCH, J. R.; SILLA, L.M.R. Prevalence of
thalassemias and variant hemoglobin in patients with non-ferropenic anemia. Rev. Bras. Hematol. Hemoter., v.27, n.1, p.37-42, 2005.
WEATHERALL, D.J. Phenotype-genotype relationships in
monogenic disease: lessons from the thalassaemias. Nat. Rev. Genet., v.2, n.4, p.245-255, 2001.
WENNING, M.R.S.C.; KIMURA, E.M.; COSTA, F.F.; SAAD, S.T.O.; GERVÁSIO, S.; JORGE, S.B.; BORGES, E.; SILVA, n.M.; SONATI, M.F. α-globin genes: thalassemic
and structural alterations in a Brazilian population. Braz. J. Med. Biol. Res., v.33, n.9, p.1041-1045, 2000.