Dissertação de Mestrado
2.º Ciclo em Biotecnologia para as Ciências da Saúde
O MLPA (Multiplex Ligation-dependent Probe Amplification)
e o aCGH (array Comparative Genomic Hybridization) no
diagnóstico de alterações cromossómicas
ANA KARINA DA SILVA MENDES
Orientadora: Professora Doutora Isabel Marques Carreira
Coorientador: Professor Doutor Gilberto Igrejas
UNIVERSIDADE DE TRÁS-OS-MONTES E ALTO DOURO VILA REAL, 2012
Pá gin a
i
Júri de Apreciação
Presidente –
.
1º Vogal –
.
2º Vogal –
.
3º Vogal –
.
Classificação:
.
Data: / /
.
Pá
gin
a
Pá
gin
a
iii
“A vida é uma muito complexa estrutura de informação e todas as suas atividades envolvem processamento de informação”
Pá
gin
a
Pá
gin
a
v
Agradecimentos
Ao finalizar este trabalho, não posso deixar de agradecer a um conjunto de pessoas que direta ou indiretamente, me apoiaram e motivaram na elaboração desta Tese, e assim, a concluir mais uma etapa da minha formação académica.
À Universidade de Trás-os-Montes e Alto Douro em nome do Professor Doutor Carlos Alberto Sequeira, Reitor da UTAD, pelas facilidades concedidas na realização deste trabalho.
Ao Diretor da Faculdade de Medicina da Universidade de Coimbra, em nome do Professor Doutor Manuel Santos Rosa, por consentir a realização deste trabalho experimental, nesta instituição.
Aos Docentes do 1º e 2º ciclo, que ao longo do Curso e Mestrado me transmitiram os seus conhecimentos e contribuíram assim para o enriquecimento da minha formação académica e científica e da mesma forma me ajudaram a perspetivar acerca dos meus interesses futuros.
Ao Coordenador do Mestrado em Biotecnologia para as Ciências da Saúde, Professor Doutor Valdemar Carnide, agradeço a oportunidade que tive em frequentar este Mestrado.
À Professora Doutora Isabel Carreira, orientadora desta Tese, quero agradecer a oportunidade que me foi dada de integrar durantes estes meses a equipa do seu laboratório, o qual foi fundamental para aprofundar e pôr em prática os meus conhecimentos nesta área. Da mesma forma, não posso deixar de mostrar o meu sincero e profundo agradecimento pela sua orientação, amabilidade e disponibilidade.
Ao Professor Doutor Gilberto Igrejas, coorientador desta Tese, por todo o apoio, profissionalismo e disponibilidade que sempre demonstrou ao longo deste trabalho.
À Professora Doutora Joana Barbosa de Melo, o meu sincero agradecimento por toda a colaboração, disponibilidade e apoio científico na elaboração desta Tese.
Ao Laboratório de Citogenética e Genómica da Faculdade de Medicina da Universidade de Coimbra, onde foi realizado este trabalho, agradeço a toda a Equipa do Laboratório, o apoio, a simpatia e a disponibilidade, em especial, ao José Ferrão, ao Miguel Pires e à Susana Ferreira, os quais estiveram de forma mais próxima envolvidos na realização prática deste trabalho.
Expresso também a minha gratidão e solidariedade a Todos os Pacientes, que prestaram uma contribuição fundamental para que este trabalho fosse possível.
Pá
gin
a
vi
Às Minhas Amigas e Colegas de Estágio, Cátia e Diana, e também à Ilda, um muito obrigado pelo apoio, amizade, boa disposição e pela partilha de bons momentos, o qual foi fundamental para a minha integração em Coimbra.
Aos Meus Amigos, por todo o convívio, amizade e companheirismo e em particular ao Leandro que sempre me apoiou e incentivou ao longo desta etapa da minha vida.
À Minha Família, em especial aos Meus Pais e aos Meus Irmãos, por todo o apoio, carinho, compreensão e em particular todo o incentivo que sempre me deram e continuam a dar nas minhas decisões, o qual me dá força para seguir em frente e alcançar os meus objetivos. A eles, dedico todo este trabalho.
Pá
gin
a
vii
Resumo
Com a introdução das novas tecnologias de biologia molecular em diagnóstico citogenético-genómico, desequilíbrios cromossómicos inferiores a 3-5 Mb, que outrora não seriam identificados pela citogenética convencional, passaram a ser detetados. De entre essas tecnologias, destacam-se as técnicas de MLPA e aCGH utilizadas neste trabalho e com a capacidade de detetarem rearranjos estruturais desequilibrados. O aCGH identifica desequilíbrios ao nível de todo o genoma do paciente num único ensaio e possibilita a determinação exata do tamanho do intervalo deletado ou amplificado, incluindo o número de genes envolvidos. Por sua vez, o MLPA permite o estudo direcionado de regiões cromossómicas, para as quais existe um número limitado de sondas a cobrir a região (na maioria das vezes o tamanho real do rearranjo cromossómico é subestimado devido às distâncias entre as sondas de MLPA, que podem encobrir uma deleção/duplicação).
Durante o período em que decorreu este trabalho, no âmbito do diagnóstico pré-natal foram recebidas no Laboratório de Citogenética e Genómica 147 amostras (sobretudo fetos com malformações ecográficas e/ou rastreio bioquímico positivo), para despiste das aneuploidias mais comuns (13, 18, 21, X e Y) através da técnica de MLPA. Destas, estudamos neste trabalho 89 amostras através do painel de MLPA P095, específico para as aneuploidias mais comuns. No âmbito do diagnóstico pós-natal utilizamos 5 painéis de MLPA, direcionados para o estudo de regiões cromossómicas específicas, tendo sido analisados 24 pacientes com o painel P250 (síndrome de DiGeorge), 19 pacientes com os painéis P036 e P070 (regiões subteloméricas), 18 pacientes com o painel P245 (síndromes de microdeleção/microduplicação) e 16 pacientes com o painel P343 (autismo). Em relação à técnica de aCGH (4X180K) dos 230 pacientes estudados neste laboratório durante o período a que se refere a este estudo, selecionamos para discussão neste trabalho 3 pacientes com alterações cromossómicas específicas.
Em termos de resultados, o painel P095 utilizado para a pesquisa de aneuploidias foi o que apresentou um maior número de amostras analisadas comparativamente aos restantes painéis de MLPA utilizados no âmbito do diagnóstico pós-natal. Esta situação deve-se ao facto de, atualmente, a maioria dos estudos em pós-natal estar centralizada na técnica de aCGH com a exceção do painel P250 (síndrome de DiGeorge), o qual permanece como a estratégia inicial no estudo de pacientes com anomalias cardíacas como indicação clínica principal, devido à elevada densidade de sondas específicas para a região crítica da síndrome de DiGeorge localizada no cromossoma 22q11.21. Em termos de diagnóstico pós-natal, através das técnicas de MLPA e aCGH analisamos seis alterações cromossómicas em pacientes com quadros clínicos específicos, as quais foram posteriormente confirmadas pelas técnicas de MLPA ou de FISH. Dessas
Pá
gin
a
viii
alterações cromossómicas observadas, duas são de novo, uma herdada e as restantes de origem desconhecida.
Ambas as tecnologias utilizadas neste trabalho foram fundamentais para o estudo de pacientes com fenótipos particulares, tendo sido determinada na maioria dos casos uma correlação genótipo-fenótipo para as anomalias cromossómicas identificadas.
Palavras-chave: MLPA; aCGH; FISH; diagnóstico; deleções; duplicações; aneuploidias.
Pá
gin
a
ix
The MLPA (Multiplex Ligation-dependent Probe Amplification) and aCGH (array Comparative Genomic Hybridization) in the diagnosis of chromosomal aberrations
Abstract
With the introduction of new molecular biology technologies in the cytogenetic-genomic diagnosis, chromosomal imbalances lower than 3-5 Mb, which formerly would not be identified by conventional cytogenetic, began to be detected. Among those technologies, highlights are the MLPA and aCGH techniques used in this study and with the capacity to detect unbalanced structural rearrangements. The aCGH technique identifies imbalances in the entire genome of the patient in a single assay and allows the determination of the exact size of the gap deleted or amplified, including the number of genes involved. In turn, the MLPA technique allows the targeted study of chromosomal regions, for which there is a limited number of probes covering the region (in most cases, the real size of the chromosomal rearrangement is underestimated due to the distances between the MLPA probes, which may mask a deletion/duplication).
During the period in which this study took place, in the context of prenatal diagnosis were received, at the Laboratory of Cytogenetics and Genomics, 147 samples (especially from fetuses with sonographic abnormalities and/or positive biochemical screening), for the screening of the most common aneuploidies (13, 18, 21, X and Y) using the MLPA technique. Of these, 89 samples were studied using the P095 panel, specific for the most common aneuploidies. In the context of postnatal diagnosis we used 5 MLPA panels, targeted to the study of specific chromosomal regions. 24 patients were analysed with the P250 panel (DiGeorge syndrome), 19 patients with the P036 and P070 panels (subtelomeric regions), 18 patients with P245 panel (microdeletion/microduplication syndromes), and 16 patients with P343 panel (autism). Regarding the aCGH (4X180K) technique, of the 230 patients studied in this laboratory during the period referred to this study, we selected for discussion in this work 3 patients with specific chromosomal alterations.
In terms of results, the P095 panel, used for the detection of aneuploidies in prenatal studies, presented the greater number of samples compared to the other panels used in the postnatal diagnosis. This is due to the fact that, currently, most studies on postnatal are centered in the aCGH technique with the exception of panel P250 (DiGeorge syndrome), which remains as the initial strategy in the study of patients presenting heart abnormalities as main clinical indication, because of the high density of probes specific for the critical region of DiGeorge syndrome located on chromosome 22q11.21. In terms of postnatal diagnosis, through the techniques of MLPA and aCGH we have identified six chromosomal abnormalities in patients
Pá
gin
a
x
with specific clinical conditions, which were subsequently confirmed by MLPA or FISH techniques. Of the observed chromosomal alterations, two are de novo, one inherited and the remaining of unknown origin.
Both technologies used in this study were fundamental to the study of patients with particular phenotypes, having been determined in most cases a genotype-phenotype correlation for the chromosomal abnormalities identified.
Key-words: MLPA; aCGH; FISH; diagnosis; deletions; duplications; aneuploidies.
Pá gin a
xi
Índice geral
Agradecimentos ... v Resumo ... viiResumo Gráfico ... viii
Abstract ... ix
Graphical abstract ... x
Lista de Símbolos e Abreviaturas ... xxi
1. Introdução Teórica ... 1
1.1. Diagnóstico citogenético-genómico ... 1
1.1.1. Diagnóstico pré-natal ... 1
1.1.2. Técnicas de DPN não invasivas ... 2
1.1.2.1. Ecografia fetal ... 3
1.1.2.2. Imagem de ressonância magnética... 3
1.1.2.3. Teste bioquímico (marcadores do soro materno) ... 3
1.1.2.4. Cell-free fetal DNA em DPN ... 4
1.1.3. Técnicas de DPN invasivas ... 4
1.1.3.1. Biópsia do trofoblasto ... 5
1.1.3.2. Amniocentese... 6
1.1.3.3. Cordocentese ... 7
1.1.3.4. Biópsia de tecidos fetais ... 8
1.1.4. Diagnóstico pós-natal ... 8
1.2. Alterações cromossómicas detetadas em diagnóstico citogenético-genómico ... 12
1.2.1. Alterações cromossómicas numéricas ... 12
1.2.2. Alterações cromossómicas estruturais ... 13
1.2.2.1. Translocações... 13
1.2.2.2. Inversões ... 14
1.2.2.3. Inserções ... 15
Pá gin a
xii
1.2.2.5. Duplicações ... 16 1.2.2.6. Isocromossomas ... 17 1.2.2.7. Cromossomas dicêntricos ... 17 1.2.2.8. Cromossomas em anel ... 18 1.2.2.9. Cromossomas marcadores... 19 1.2.3. Mosaicismo ... 191.3. Técnicas usualmente utilizadas em diagnóstico citogenético-genómico ... 20
1.3.1. Citogenética convencional ... 20
1.3.2. Citogenética molecular e genómica ... 21
1.3.2.1. FISH (Fluorescence in situ Hybridization) ... 22
1.3.2.1.1. Principais sondas usadas na técnica de FISH ... 23
1.3.2.1.2. Vantagens e limitações da técnica de FISH ... 24
1.3.2.2. MLPA (Multiplex Ligation-dependent Probe Amplification) ... 24
1.3.2.2.1. Vantagens e limitações da técnica de MLPA... 26
1.3.2.3. aCGH (array Comparative Genomic Hybridization) ... 27
1.3.2.3.1. Vantagens e limitações da técnica de aCGH ... 29
1.3.2.3.2. CNVs e a sua importância em diagnóstico ... 30
2. Objetivos ... 33
3. Material e Métodos ... 35
3.1. Amostras biológicas ... 35
3.1.1. Diagnóstico Pré-natal ... 35
3.1.2. Diagnóstico Pós-natal ... 35
3.2. Extração de DNA a partir de amostras de biópsia de vilosidades coriónicas, de líquido amniótico e de sangue periférico ... 36
3.3. Quantificação do DNA ... 38
3.4. Aplicação da técnica de MLPA ... 38
3.5. Aplicação da técnica de FISH ... 41
3.6. Aplicação da técnica de array CGH ... 44
4. Resultados e Análise ... 47
Pá
gin
a
xiii
4.1.1. Interpretação dos eletroferogramas ... 47
4.2. Diagnóstico pré-natal ... 48
4.2.1. Aneuploidias (Painel P095) ... 49
4.3. Diagnóstico pós-natal ... 50
4.3.1. Região crítica 22q11.21 (Painel P250) ... 50
4.3.1.1. Deleção na região crítica 22q11.21 ... 51
4.3.1.2. Duplicação na região crítica 22q11.21 ... 53
4.3.2. Estudo das regiões subteloméricas (Painéis P036 e P070) ... 54
4.3.2.1. Duplicação na região X/Yqter ... 54
4.3.3. Deleção em mosaico na região 2p14 (aCGH) ... 56
4.3.4. Deleção na região 15q13.2q13.3 (aCGH) ... 59
4.3.5. Duplicação na região crítica 7q11.23 (aCGH) ... 60
5. Discussão ... 63
5.1. Aneuploidias (Painel P095) ... 63
5.1.1. Aneuploidias autossómicas (trissomias 21, 18 e 13) ... 63
5.1.2. Aneuploidias dos cromossomas sexuais (cromossomas X e Y) ... 64
5.1.3. Causa das aneuploidias ... 65
5.1.4. Técnicas de DPN invasivas e amostras analisadas (%) ... 66
5.2. Síndromes de microdeleção/microduplicação (Painel P245)... 67
5.3. Autismo (Painel P343) ... 67
5.4. Região crítica 22q11.21 (Painel P250) ... 68
5.4.1. Síndrome de DiGeorge/Velocardiofacial ... 68
5.4.2. Mecanismo de formação ... 71
5.4.3. Correlação genótipo-fenótipo ... 72
5.4.3.1. Deleção na região crítica 22q11.21 ... 73
5.4.3.2. Duplicação na região crítica 22q11.21 ... 74
5.5. Duplicação na região X/Yqter (Painéis P036 e P070) ... 75
5.5.1. Alterações nas regiões subteloméricas e mecanismos de formação ... 75
Pá
gin
a
xiv
5.5.3. Correlação genótipo-fenótipo ... 77
5.6. Deleção em mosaico na região 2p14 (aCGH) ... 78
5.6.1. Mecanismo de formação ... 79
5.6.2. Correlação genótipo-fenótipo ... 80
5.7. Deleção na região 15q13.2q13.3 (aCGH) ... 82
5.7.1. Principais síndromes associadas ao cromossoma 15q ... 83
5.7.2. Mecanismo de formação ... 84
5.7.3. Correlação genótipo-fenótipo ... 85
5.8. Duplicação na região crítica 7q11.23 (aCGH) ... 86
5.8.1. Síndrome de Williams-Beuren ... 86
5.8.2. Síndrome de duplicação 7q11.23 ... 87
5.8.3. Mecanismo de formação ... 88
5.8.4. Correlação genótipo-fenótipo ... 89
6. Conclusões e perspetivas futuras ... 91
7. Referências Bibliográficas... 95
8. Anexos ... 107
Índice de Figuras
Figura 1.1 – Amostra de células do trofoblasto obtida transabdominalmente, com auxílio de uma sonda ecográfica e de uma agulha [adaptado de (Binns & Hsu 2001)]. ... 5Figura 1.2 – DPN por amniocentese. Com auxílio de uma sonda ecográfica e de uma agulha é retirado LA, a partir do qual podem ser efetuadas inúmeras análises, por exemplo, estudos de DNA [adaptado de (Binns & Hsu 2001)]. ... 7
Figura 1.3 – Cordocentese em DPN. Com o auxílio de uma sonda ecográfica e de uma agulha é retirado sangue fetal a partir do cordão umbilical [adaptado de (Binns & Hsu 2001)]. ... 8
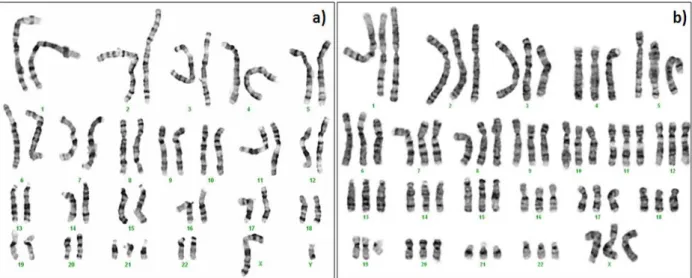
Figura 1.4 – Alterações cromossómicas numéricas: a) trissomia 21 resultante de um cromossoma 21 extra (47,XY,+21); b) triploidia (3n) com um total de 69 cromossomas (69,XXX) (imagem cedida pelo laboratório de Citogenética e Genómica, Faculdade de Medicina da Universidade de Coimbra). ... 13
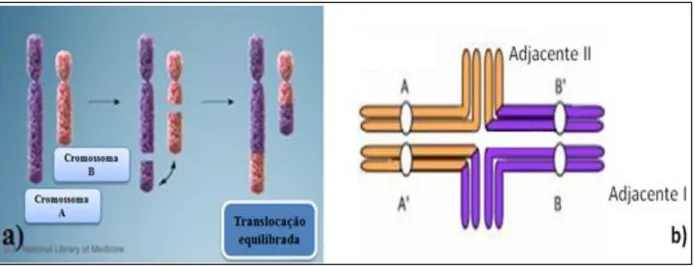
Figura 1.5 – a) Numa translocação equilibrada, os fragmentos cromossómicos são rearranjados sem perda ou ganho de material genético na célula (adaptado de
Pá
gin
a
xv
http://ghr.nlm.nih.gov/handbook/illustrations); b) Emparelhamento durante o paquíteno de
cromossomas com uma translocação recíproca (adaptado de
http://www.icb.ufmg.br/big/genegrad/genetica/img/CromossMutaTermno_clip_image016.jpg). ... 14
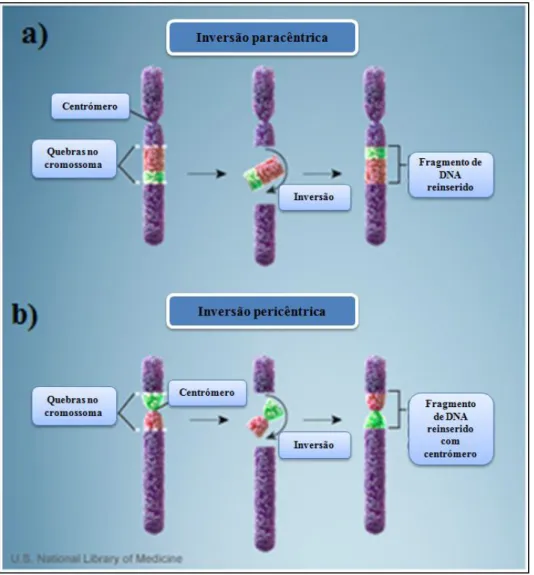
Figura 1.6 – As inversões envolvem duas quebras num cromossoma e o fragmento de DNA resultante é invertido e reinserido no cromossoma: a) inversão paracêntrica (não envolve o centrómero); b) inversão pericêntrica (envolve o centrómero) (adaptado de http://ghr.nlm.nih.gov/handbook/illustrations). ... 15
Figura 1.7 – Inserção (adaptado de
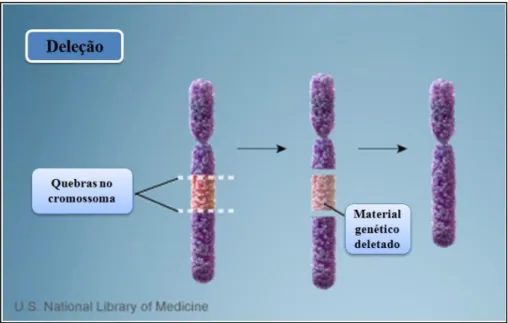
http://mset.rst2.edu/portfolios/s/saffran_m/thesis/insertion.jpg). ... 15 Figura 1.8 – Numa deleção ocorrem quebras num cromossoma e perda do material genético entre as mesmas (adaptado de http://ghr.nlm.nih.gov/handbook/illustrations). ... 16
Figura 1.9 – Numa duplicação parte de um cromossoma é copiada (duplicada), resultando em material genético extra a partir do segmento duplicado (adaptado de http://ghr.nlm.nih.gov/handbook/illustrations). ... 16
Figura 1.10 – Divisão transversal de um cromossoma e formação de um cromossoma anormal com dois braços idênticos, dois braços curtos ou dois braços longos, o qual é designado de isocromossoma (adaptado de http://ghr.nlm.nih.gov/handbook/illustrations). ... 17
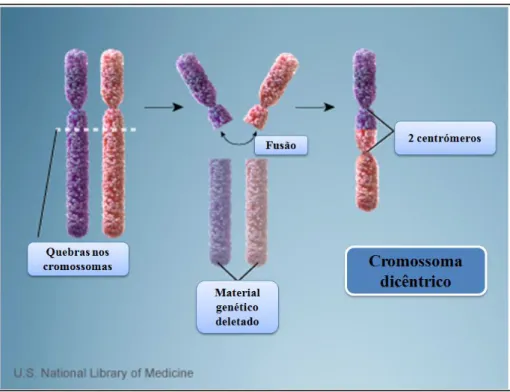
Figura 1.11 – Formação de um cromossoma dicêntrico. Este mecanismo envolve quebras em dois cromossomas e fusão dos fragmentos resultantes, cada um deles incluindo um centrómero (adaptado de http://ghr.nlm.nih.gov/handbook/illustrations). ... 18
Figura 1.12 – Formação de um cromossoma em anel. Este mecanismo envolve quebras em dois sítios de um cromossoma e fusão de ambas as extremidades resultantes para formar uma estrutura circular com perda dos fragmentos acêntricos (adaptado de http://ghr.nlm.nih.gov/handbook/illustrations). ... 18
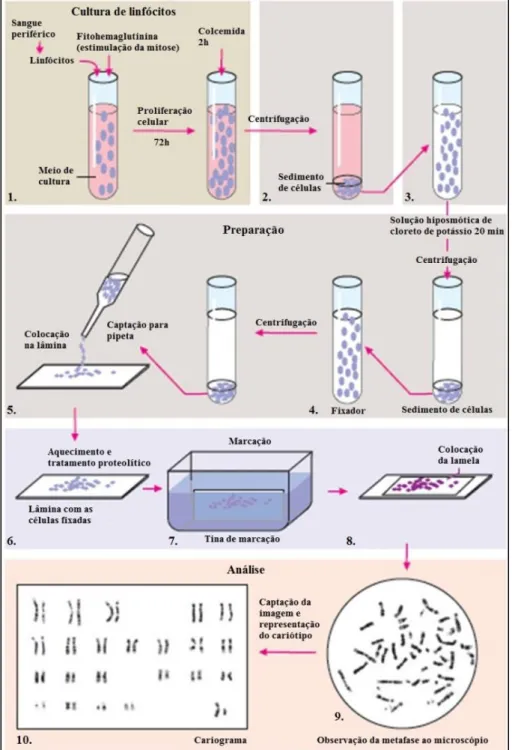
Figura 1.13 – Principais etapas envolvidas na obtenção de um cariograma a partir de amostras de sangue periférico: cultura de linfócitos (1), colheita dos cromossomas em metafase (2), preparação dos cromossomas (3-5), bandagem (6-8), análise das metafases ao microscópio (9) e determinação do cariograma (10) [adaptado de (Passarge 2007)]. ... 21
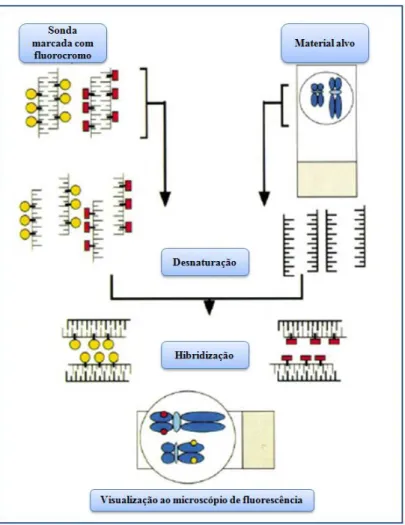
Figura 1.14 – Princípio da técnica de FISH. As sondas de FISH marcadas com fluorescência e os cromossomas em metafase são sujeitos a um processo de desnaturação. Posteriormente ocorre a hibridização entre as sondas de FISH e o DNA alvo, sendo a visualização dos respetivos sinais de fluorescência realizada ao microscópio de fluorescência [adaptado de (Montironi et al. 2003)]. ... 23
Pá
gin
a
xv
i
Figura 1.15 – Principais etapas envolvidas numa reação de MLPA. Durante a primeira etapa, o DNA é desnaturado e incubado com uma mistura de sondas de MLPA. Cada sonda de MLPA consiste em dois oligonucleótidos individuais, em que cada um possui uma das sequências primer PCR. Estas duas sondas de oligonucleótidos hibridizam nas sequências alvo adjacentes (1) e por conseguinte são ligadas durante a reação de ligação (2). Após a ligação ocorre a PCR, no qual apenas as sondas ligadas irão ser amplificadas exponencialmente com um par de primers universal (3). Numa última etapa os produtos de amplificação são separados usando a eletroforese capilar (4) (adaptado de http://www.mlpa.com). ... 26
Figura 1.16 – Princípio da técnica CGH. O DNA do paciente e o DNA da amostra controlo são marcados diferencialmente com Cy3 (verde) e Cy5 (vermelho) e co-hibridizam em metafases normais. A avaliação das diferenças na intensidade da fluorescência permite detetar alterações no número de cópias, as quais são representadas em cariótipo [adaptado de (Tönnies 2002)]. ... 27
Figura 1.17 – Etapas na obtenção de perfis de aCGH. O DNA do paciente e o DNA do controlo são marcados diferencialmente com Cy3 e Cy5 e hibridizam competitivamente num microarray genómico. O array consiste em DNAs alvo selecionados, que abrangem regiões cromossómicas ou o genoma completo. A razão entre as intensidades dos dois sinais de fluorescência reflete o número relativo de cópias, sendo o rácio para cada spot disposto em relação à sua posição correspondente no genoma humano para dar origem ao perfil do número de cópias [adaptado de (Chari et al. 2006)]. ... 28
Figura 3.1 – Esquema ilustrativo da estratégia utilizada na interpretação de alterações detetadas pelas técnicas de MLPA ou aCGH no âmbito de estudos em pós-natal. ... 36
Figura 3.2 – Etapas essenciais no processo de extração de DNA com kits comerciais. ... 37 Figura 3.3 – Esquema ilustrativo das principais etapas e respetivos equipamentos utilizados num ensaio de MLPA. ... 40
Figura 3.4 – Etapas principais num estudo de FISH: pré-tratamento, desnaturação, hibridização, lavagem, montagem e visualização ao microscópio de fluorescência (adaptado de http://www.abnova.com/support/FISH.asp). ... 42
Figura 3.5 – Principais etapas envolvidas numa experiência de aCGH. ... 44 Figura 4.1 – Eletroferograma de uma amostra analisada com o painel P250-B1. Os picos de fluorescência iniciais dizem respeito aos fragmentos controlo da quantidade (Q) e desnaturação (D) do DNA (incluindo os fragmentos específicos dos cromossomas X e Y). Os restantes picos de fluorescência representam cada uma das 48 sondas de MLPA presentes neste painel, as quais estão associadas a genes específicos tal como podemos visualizar na imagem apresentada. ... 48
Pá
gin
a
xv
ii
Figura 4.2 – A) Percentagem dos diferentes tipos de amostras analisadas em DPN. B) Percentagem das diferentes aneuploidias observadas. ... 49
Figura 4.3 – Resultado obtido a partir da análise com o painel P095-A3. A média da área relativa obtida para cada um dos cromossomas (13, 18, 21 e X) é dividida individualmente pela média da área relativa de cada um dos outros cromossomas em análise. A partir desta divisão é obtido um rácio que nos indica se a amostra apresenta uma trissomia (valores superiores a 1,3), uma monossomia (valores inferiores a 0,7) ou então se é normal (valores próximos de 1,0). Neste caso específico, foi identificada uma trissomia do cromossoma 21, num indivíduo do sexo feminino. ... 50
Figura 4.4 – Resultado da FISH obtido com as sondas N25 e N85A3 (sonda controlo). Para a região crítica da síndrome de DiGeorge em 22q11.21 (sonda N25) não foi detetado qualquer rearranjo, como indicado pelas setas. ... 51
Figura 4.5 – Resultado obtido com o painel de MLPA P250-B1. Como se pode observar a partir desta imagem, o probando e o progenitor apresentam a mesma deleção (valores inferiores a 0,7) na região crítica do cromossoma 22q11.21, com 5 sondas de MLPA alteradas (ZNF74 a LZTR1), sendo a progenitora normal para esta região cromossómica. ... 52
Figura 4.6 – Resultado obtido com o painel de MLPA P250-B1. Após o estudo dos irmãos do probando e como se pode observar pela figura apresentada, ambos revelaram-se normais para a região crítica do cromossoma 22q11.21 (valores próximos de 1,0). ... 52
Figura 4.7 – Resultado obtido com o painel de MLPA P250-B1. De acordo com esta imagem, a primeira amostra representa o controlo positivo para a região tipicamente duplicada em pacientes com a síndrome de duplicação 22q11.21 (região com cerca de 3 Mb). A segunda amostra refere-se ao probando, o qual apresenta uma duplicação (valores superiores a 1,3) na região crítica do cromossoma 22q11.21, com 5 sondas alteradas (ZNF74 a LZTR1). ... 53
Figura 4.8 – Resultado obtido com o painel de MLPA P036-E1. Como se pode visualizar a partir desta figura, apenas o probando apresenta uma duplicação (valor superior a 1,3) na região X/Yqter, sendo os progenitores e o irmão normais para esta região (valor próximo de 1,0). ... 54 Figura 4.9 – Resultado obtido com o painel de MLPA P070-B1. Como se pode visualizar a partir desta figura, apenas o probando apresenta uma duplicação (valor superior a 1,3) na região X/Yqter, sendo os progenitores e o irmão normais para esta região (valor próximo de 1,0). ... 55 Figura 4.10 – Resultado da FISH obtido com as sondas ToTelVysion 2p (VIJ yRM2052) 2q (D2S447) e Xq/Yq (EST Cdy 16c07). Tal como indicado na figura a duplicação localiza-se no cromossoma Y. ... 55
Pá
gin
a
xv
iii
Figura 4.11 – Resultado da FISH obtido com a sonda ToTelVysion Xq/Yq (EST Cdy 16c07). Tal como indicado na figura não existe qualquer translocação envolvendo a região X/Yqter... 56
Figura 4.12 – Representação esquemática do tamanho da região 2p14 deletada (a azul) e ideograma do cromossoma 2 mostrando a vermelho a localização dessa mesma região (adaptado da base de dados genómica UCSC, February 2009, http://genome.ucsc.edu). ... 56
Figura 4.13 – Resultado obtido pela técnica de aCGH, onde se pode observar entre o probando marcado com Cy5 (vermelho) e o controlo com Cy3 (verde) uma deleção na região 2p14. A) Perfil para o cromossoma 2, com a deleção assinalada; B) Conteúdo génico da deleção. ... 57 Figura 4.14 – Resultado da FISH obtido com a sonda RP11-568N6, a qual representa a região 2p14. Como indicado pelas setas (e núcleo interfásico), ambos os cromossomas 2 apresentam sinais vermelhos fluorescentes (sem deleção na região 2p14). ... 57
Figura 4.15 – Resultado da FISH obtido com a sonda RP11-568N6, a qual representa a região 2p14. A) Como indicado pelas setas o sinal fluorescente foi apenas detetado num dos cromossomas 2 (deleção na região 2p14). B) A partir das setas são visualizados dois sinais fluorescentes correspondentes a ambos os cromossomas 2 (sem deleção na região 2p14). ... 58
Figura 4.16 – Resultado da FISH obtido com a sonda RP11-568N6, a qual representa a região 2p14. A) Como indicado pelas setas, na progenitora foram identificados sinais vermelhos fluorescentes em ambos os cromossomas 2 (sem deleção na região 2p14). B) No caso do progenitor verifica-se a mesma situação (sem deleção na região 2p14). ... 58
Figura 4.17 – Representação esquemática do tamanho da região 15q13.2q13.3 deletada (a azul) e ideograma do cromossoma 15 mostrando a vermelho a localização dessa mesma região (adaptado da base de dados genómica UCSC, February 2009, http://genome.ucsc.edu). ... 59
Figura 4.18 – Resultado obtido pela técnica de aCGH, onde se pode observar entre o probando marcado com Cy5 (vermelho) e o controlo com Cy3 (verde) uma deleção na região 15q13.2q13.3. A) Perfil para o cromossoma 15, com a deleção assinalada; B) Conteúdo génico da deleção. ... 59
Figura 4.19 – Resultado obtido com o painel de MLPA P343-C1. Tal como se pode visualizar nesta figura, a primeira amostra diz respeito ao controlo positivo da deleção na região 15q13.3. A segunda amostra é a do probando, onde tal como no caso do controlo positivo podemos observar uma deleção (valores inferiores a 0,7) na região 15q13.3, com 3 sondas alteradas. ... 60
Figura 4.20 – Resultado obtido pela técnica de aCGH, onde se pode observar entre o probando marcado com Cy5 (vermelho) e o controlo com Cy3 (verde) uma duplicação na região
Pá
gin
a
xix
crítica do cromossoma 7q11.23. A) Perfil para o cromossoma 7, com a duplicação assinalada; B) Conteúdo génico da duplicação. ... 61
Figura 4.21 – Representação esquemática do tamanho da região 7q11.23 duplicada (a azul) e ideograma do cromossoma 7 mostrando a vermelho a localização dessa mesma região (adaptado da base de dados genómica UCSC, February 2009, http://genome.ucsc.edu). ... 61
Figura 4.22 – Resultado obtido com o painel de MLPA P245-A2. Tal como podemos observar a partir desta imagem, o probando apresenta uma duplicação (valores superiores a 1,3) na região 7q11.23 com duas sondas alteradas, enquanto que, o irmão é normal para esta região (valores próximos de 1,0). ... 62
Figura 5.1 – A) NAHR intercromossómica pode originar deleções e duplicações. B) NAHR intercromatídea pode também levar a deleções e duplicações. C) NAHR intracromatídea resulta em deleções e em cromossomas em anel. D) NAHR intracromatídea entre LCRs em orientação oposta pode resultar em inversão [adaptado de (Colnaghi et al. 2011)]. ... 72
Figura 5.2 – Representação esquemática do mapa físico da região crítica do cromossoma 22q11.21, onde podem ser visualizadas as localizações dos LCR22s e a maioria dos pontos de quebra nas síndromes de deleção e duplicação nessa região [adaptado de (Portnoї 2009)]. ... 72
Figura 5.3 – Ideograma do cromossoma 15q, onde se pode observar a região crítica das síndromes de Prader-Willi/Angelman (PWS/AS) numa posição proximal (15q11q14) e a região 15q13.2q13.3 entre os LCRs BP4 e BP5 numa região mais distal [adaptado de (Miller et al. 2009)]. ... 85
Figura 5.4 – Representação esquemática dos principais rearranjos genómicos na região crítica do cromossoma 7q11.23. Os LCRs centromérico (c), mediano (m) e telomérico (t) são representados pelas setas coloridas, onde é também evidente o sentido de orientação entre os mesmos [adaptado de (Merla et al. 2010)]. ... 88
Figura 8.1 – Representação esquemática da região Yq12 (gene VAMP7 ou SYBL1) de acordo com a base de dados DGV, onde a barra a azul corresponde a ganhos nesta região cromossómica descritos por Perry e colaboradores em 2008. ... 107
Figura 8.2 – Representação esquemática de acordo com a base de dados DECHIPHER, onde as barras a vermelho correspondem a pacientes com deleção e as barras a azul a pacientes com duplicação. A partir desta figura podem ser observados 3 pacientes com deleção de dimensão superior à deleção na região 2p14 (delimitada pelo retângulo preto) identificada no probando. ... 107
Figura 8.3 – Representação esquemática de acordo com a base de dados DECHIPHER, onde as barras a vermelho correspondem a pacientes com deleção e as barras a azul a pacientes com duplicação. A região 15q13.2q13.3 deletada no probando corresponde à região delimitada
Pá
gin
a
xx
pelo retângulo preto, na qual existem diversos pacientes com deleção (ou duplicação) sobreposta a essa região. ... 108
Índice de Quadros
Quadro 1.1 – Síndromes de microdeleção/microduplicação e gene/locus envolvido (s) em cada uma delas, com a respetiva localização cromossómica [adaptado de (Theisen & Shaffer 2010)]. ... 11
Quadro 4.1 – Total de amostras analisadas através dos painéis de MLPA P095-A3 (aneuploidias mais comuns), P250-B1 (síndrome de DiGeorge), P036-E1 e P070-B1 (regiões subteloméricas), P245-A2 (síndromes de microdeleção/microduplicação) e P343-C1 (autismo) (MRC-Holland, Amsterdam, Netherlands) e número de alterações cromossómicas identificadas em cada um desses painéis. ... 47
Quadro 4.2 – Número de pacientes estudados em cada um dos tipos de amostras e aneuploidias detetadas... 49
Pá
gin
a
xxi
Lista de Símbolos e Abreviaturas
Símbolos
% - Percentagem
°C - Graus Celsius
A
aCGH - array Comparative Genomic
Hybridization
ACTR2 - ARP2 actin-related protein 2
homolog (yeast)
ADAM12 - A Disintegrin Metalloprotease 12
ADM-2 - Aberration Detection Method-2
ADPM - Atraso no Desenvolvimento Psicomotor
ARHGAP11B - Rho GTPase activating protein 11B
ASD - Autism Spectrum Disorder
B
BAC - Bacterial artificial chromosome
BAZ1B - bromodomain adjacent to zinc
finger domain, 1B BP1-BP3 - breakpoint 1- breakpoint 3 BP2-BP3 - breakpoint 2- breakpoint 3 BP4 breakpoint 4 BP5 - breakpoint 5
C
cDNA - complementary Deoxyribonucleic
acid
CEP - centrosomal protein
CEP68 - centrosomal protein 68kDa
cffDNA - cell-free fetal Deoxyribonucleic
acid
CGH - Comparative Genomic
Hybridization
CHRNA7 - cholinergic receptor, nicotinic,
alpha 7 (neuronal)
CNVs - Copy-number variations
CRKL - v-crk sarcoma virus CT10 oncogene
homolog (avian)-like
CVS - Chorionic Villus sampling
CXYorf1 - CXYorf1 protein
Cy3 - Cianina-3
Cy5 - Cianina-5
CYLN2 - cytoplasmic linker 2
D
DAPI - 4',6-diamidino-2-phenylindole
DECHIPHER -
Database of Chromosomal Imbalance and Phenotype in Humans Using Ensembl Resources
DGV - Database of Genomic Variants
dH2O - Água destilada
dn - de novo
DNA - Deoxyribonucleic acid
dNTPs - deoxyribonucleotide triphosphates
DPN - Diagnóstico Pré-Natal
DSB - Double Strand Break
E
Pá gin a
xx
ii
F
FAN1 - FANCD2/FANCI-associated nuclease 1FISH - Fluorescent in situ Hybridization
FoSTeS/MMBIR -
Fork Stalling and Template Switching/Microhomology Mediated
Break Induced Replication
G
GABRB3 - gamma-aminobutyric acid (GABA)
A receptor, beta 3
GTF2I - general transcription factor Iii
GTF2IRD1 - GTF2I repeat domain containing 1
I
i - Isocromossoma
IHC - Immunohistochemistry
IL9R - interleukin 9 receptor
K
kb - kilobase kDa - kilodalton KLF13 - Kruppel-like factor 13 KLHL22 - kelch-like 22 (Drosophila)L
LA’s - Líquidos amnióticos
LCR - Low Copy Repeat
LIMK1 - LIM domain kinase 1
LSI - Sondas específicas de locus
LOH - Loss of Heterozygosity
LZTR1 - leucine-zipper-like transcription
regulator 1
M
Mb - megabase
MED15 - mediator complex subunit 15
mg/mL - miligrama por mililitro
MIR211 - microRNA 211
mL - mililitro
MLPA - Multiplex Ligation-dependent Probe
Amplification mM MMEJ - - Milimolar Microhomology-Mediated End Joining
MRI - Magnetic Resonance Imaging
MS-MLPA -
Methylation-specific - Multiplex Ligation-dependent Probe
Amplification
MTMR10 - myotubularin related protein 10
MTMR15 - myotubularin related protein 15
N
NAHR - Non allelic homologous
recombination
ng - nanograma
NHEJ - Non-Homologous End Joining
nm - nanómetro
nt - nucleótido
O
OMIM - Online Mendelian Inheritance in
Man
Pá gin a
xx
iii
P
p - braço curto do cromossoma
PAC - Phage artificial chromosome
PAPP-A - Pregnancy Associated Plasma
Protein A
PAR1 - Pseudoautosomal Region 1
PAR2 - Pseudoautosomal Region 2
pat - paterno
pb - par de bases
PBS - Phosphate Buffered Saline
PCR - Polymerase Chain Reaction
pmol/µL - Picomoles por microlitro
PQBP1 - polyglutamine binding protein 1
PWS/AS - Prader-Willi Syndrome/Angelman
Syndrome
Q
q - braço longo do cromossoma
Q-PCR - Quantitative Polymerase Chain
Reaction
QF-PCR - Quantitative Fluorescent
Polymerase Chain Reaction
QI - Quociente de Inteligência
R
RAB1A - RAB1A, member RAS oncogene
family
RCIU - Restrição do Crescimento Intrauterino
RhD - Rhesus D antigen
RNA - Ribonucleic acid
RNase A - Ribonuclease A
RT-MLPA -
Reverse Transcription - Multiplex Ligation-dependent Probe
Amplification
S
SHANK3 - SH3 and multiple ankyrin repeat
domains 3
SLC1A4 -
solute carrier family 1 (glutamate/neutral amino acid
transporter), member4
SNAP29 - synaptosomal-associated protein,
29kDa
SNPs - Single Nucleotide Polymorphisms
SPRED2 - sprouty-related, EVH1 domain
containing 2
SPRY3 - sprouty homolog 3 (Drosophila)
SSC - Standard Saline Citrate
SYBL1/VAMP7 - synaptobrevin-like
1/vesicle-associated membrane protein 7
T
TBX1 - T-box 1
TE - Tris-EDTA (Ethylenediamine
Tetraacetic Acid)
Tris-HCl - Tris-Hydrochloride
TRPM1 - transient receptor potential cation
channel, subfamily M, member 1
U
UBE3A - ubiquitin protein ligase E3A
UCSC - University of California, Santa Cruz
Pá gin a
xx
iv
V
v/v - volume por volume
v-SNARES -
vesicle-soluble N-ethylmaleimide-sensitive factor attachment protein
receptors
VAMPs - vesicle-associated membrane
proteins
W
WBS - Williams-Beuren syndromeX
X - cromossoma XY
Y - cromossoma YYAC - Yeast artificial chromosome
Z
ZNF74 - zinc finger protein 74
β
β-HCG - β-Human Chorionic Gonadotropin
µ
µg µL - - micrograma microlitroPá gin a
1
1. Introdução Teórica
1.1.
Diagnóstico citogenético-genómico
1.1.1.
Diagnóstico pré-natal
O diagnóstico pré-natal (DPN) permite o estudo de anomalias ou doenças graves no feto ou embrião e chegar a um diagnóstico antes do seu nascimento. Em termos gerais, o DPN fornece informação genética, anatómica e fisiológica acerca do feto que pode ser usada para tomar decisões informadas e individualizadas em relação à gravidez (Goldman & Malone 2005). Embora alguns distúrbios genéticos sejam compatíveis com uma vida normal, muitos estão associados com mortalidade e atraso mental significativo. Uma vez realizada a identificação de distúrbios genéticos os pais, mediante aconselhamento genético, podem optar pela progressão ou interrupção médica da gravidez (Binns & Hsu 2001) de acordo com a gravidade das anomalias detetadas.
Uma das indicações mais comuns para DPN é a idade materna avançada definida como sendo a partir dos 35 anos, pois a partir desta idade verifica-se um equilíbrio entre o risco de perda fetal num procedimento de diagnóstico invasivo e a probabilidade de aparecimento de uma aneuploidia fetal não diagnosticada (Eisenberg & Wapner 2002). A aneuploidia está associada a uma não disjunção meiótica ou mitótica, ou ainda a atraso na migração de um cromossoma durante a anafase (lagging). É o tipo mais comum de anomalias cromossómicas clinicamente significativas que ocorre em cerca de 3 a 4% das gestações conhecidas (Teixeira & Carreira 2005). Em teoria, o fenómeno de não disjunção pode envolver qualquer um dos 23 pares de cromossomas, embora a maioria das aneuploidias resultantes estejam associadas a abortos espontâneos. Apesar do risco elevado de alterações cromossómicas numéricas associado à idade materna ser a indicação principal em DPN, os cariótipos resultantes podem também revelar alterações cromossómicas estruturais, tais como, translocações, inversões, inserções, deleções ou cromossomas em anel (Binns & Hsu 2001). Para além da idade materna avançada, existem outras indicações clínicas para DPN igualmente importantes, incluindo:
Anomalia ecográfica do feto;
Sinais ecográficos de alerta indicativos de um risco aumentado de anomalia cromossómica;
Pá
gin
a
2
Rastreio bioquímico positivo associado a um risco aumentado de anomalia cromossómica;
Pais com rearranjo cromossómico estrutural, mosaicismo ou aneuploidia dos cromossomas;
Feto ou nado-morto anterior com anomalia cromossómica;
Esclarecimento de um mosaicismo fetal detetado noutra amostra biológica desse feto; Risco de síndrome de instabilidade cromossómica.
Mulheres grávidas com maior predisposição para anomalias cromossómicas (usualmente devido a idade materna avançada, metabolitos do soro alterados, ou anomalias ecográficas detetadas no feto) podem ser sujeitas a métodos invasivos, para obtenção de amostras de vilosidades coriónicas (CVS - Chorionic Villus sampling), de líquido amniótico (LA) ou, mais raramente, de sangue fetal. O material obtido a partir dessas amostras é cultivado para a obtenção de células proliferativas, as quais são posteriormente preparadas de acordo com os procedimentos estabelecidos para a análise do cariótipo fetal (Ogilvie et al. 2005). Após a implementação do cariótipo constitucional para despiste de alterações cromossómicas no feto, surgiram outros procedimentos, principalmente os baseados nas tecnologias de biologia molecular, que proporcionaram um grande avanço nesta área, na medida em que possibilitaram uma determinação mais rápida dos resultados (num período de 24 a 48h), capacidade de automatização e maior precisão na deteção de pequenas alterações genómicas (inferiores a 5 Mb de DNA). De entre essas tecnologias e no âmbito do DPN destacam-se o QF-PCR (Quantitative Fluorescent Polymerase Chain Reaction) e o MLPA (Multiplex Ligation-dependent Probe Amplification) para a pesquisa das aneuploidias mais comuns (13, 18, 21, X e Y), embora existam outras técnicas disponíveis igualmente úteis para este propósito.
De uma forma geral, as técnicas usadas em DPN podem ser divididas em não invasivas (por exemplo, a ecografia e os testes bioquímicos) que não representam qualquer perigo para o feto e muito úteis sobretudo nos primeiros meses de gravidez, e as invasivas (biópsia do trofoblasto, amniocentese, cordocentese) as quais apresentam risco para o feto, o qual varia de acordo com o procedimento utilizado.
1.1.2.
Técnicas de DPN não invasivas
Para diagnóstico de anomalias congénitas e avaliação do risco de determinados distúrbios genéticos utilizam-se, habitualmente, os seguintes procedimentos não invasivos: ecografia fetal, imagem de ressonância magnética e teste bioquímico do soro materno (medição das enzimas indicativas no soro materno) (Agnieszka et al. 2007).
Pá
gin
a
3
1.1.2.1. Ecografia fetal
Habitualmente são realizadas durante a gravidez três ecografias de rotina obstétrica: a primeira por volta da 10.ª semana e a segunda e terceira à 22.ª e à 33.ª semana, respetivamente.
No primeiro trimestre de gravidez a ecografia fetal permite avaliar determinados parâmetros, entre os quais, a translucência da nuca e a presença ou ausência de ossos nasais (Zimmermann 2004), entre outros. As medições anormais da translucência da nuca podem estar associadas a trissomia 21, defeitos cardíacos, síndrome de Beckwith-Wiedemann, acondroplasia, síndrome de Smith-Lemli-Opitz, osteogénese imperfeita, síndrome de Nooman e com gravidez complicada pela hipertensão arterial (Agnieszka et al. 2007). Relativamente aos ossos do nariz, estes encontram-se ausentes durante o primeiro trimestre na maioria dos fetos com trissomia 21 (Zimmermann 2004).
1.1.2.2. Imagem de ressonância magnética
É usada em combinação com a ecografia fetal, usualmente aquando ou após a 18.ª semana de gestação. MRI (magnetic resonance imaging) constitui uma ferramenta ideal para exame de fetos com grandes ou complexas anomalias. Uma das vantagens da MRI comparativamente à ecografia, é o facto de esta permitir a observação de anomalias intracranianas, entre as quais, anomalias do tronco cerebral, fossa posterior e tubo neural (Collins & Impey 2012).
1.1.2.3. Teste bioquímico (marcadores do soro materno)
O screening no primeiro trimestre envolve a medição da PAPP-A (“pregnancy associated plasma protein A”), (PAPP-A no soro materno está em média reduzida em fetos com trissomia 21) e níveis de β-HCG (“β-human chorionic gonadotropin”) livre no soro materno (encontra-se aumentada no soro materno em fetos com trissomia 21) (Zimmermann 2004). Essas medições são usadas juntamente com o estudo ecográfico que, inclui a avaliação dos parâmetros, translucência da nuca e ausência/presença de ossos nasais. O screening no primeiro trimestre, usando os parâmetros referidos, é uma forma eficiente para a pesquisa de trissomias dos cromossomas 21, 18 e 13, triploidia e monossomia do cromossoma X. Este método de screening faz uma boa discriminação entre fetos normais e fetos portadores de aneuploidia (Zimmermann 2004). A taxa de deteção utilizando esses dois métodos supra citados de forma combinada é de cerca de 90% em relação à pesquisa da trissomia 21, com uma taxa de falsos positivos de 5% (Collins & Impey 2012).
A bioquímica do soro materno no segundo trimestre (entre a 14.ª e a 18.ª semana de gestação) envolve o triplo, o quádruplo (triplo screen e inibina A) e o screen integrado. O triplo
Pá
gin
a
4
screen é a medição dos níveis da α-fetoproteína, da gonadotrofina coriónica humana livre e do estriol livre no soro materno (Agnieszka et al. 2007).
1.1.2.4. Cell-free fetal DNA em DPN
Como os procedimentos invasivos apresentam um risco considerável de perda fetal, um objetivo procurado há muito tempo no DPN é o estabelecimento de testes não invasivos que permitam o teste genético pré-natal (Zimmermann 2004). Um procedimento possível é através da deteção de DNA fetal livre (cffDNA - cell-free fetal DNA) presente no plasma materno (Lo et al. 1997; Raymond et al. 2010) mas que é rapidamente eliminado após o parto (Raymond et al. 2010).
Desde a primeira descrição da presença de DNA fetal na circulação materna, em 1998, a sua amplificação por PCR emergiu como a estratégia principal para o desenvolvimento de métodos não invasivos em DPN. O cffDNA é detetável a partir da 4.ª semana de gestação e constitui cerca de 3 a 6% do DNA total livre na circulação materna nas primeiras semanas, aumentando ao longo da gestação (Collins & Impey 2012). Ao contrário do DNA celular, o cffDNA consiste predominantemente em fragmentos curtos de DNA, dos quais 80% são menores do que 193 pb (Wright & Burton 2009). Uma das principais aplicações clínicas é a previsão do sexo fetal (pelo PCR em tempo real quantitativo), em situações de gravidez com risco de distúrbios ligados ao cromossoma X (Zimmermann 2004; Collins & Impey 2012). Outras aplicações incluem a determinação do estado RhD fetal em mães RhD-negativas e DPN de algumas doenças monogénicas (por exemplo, distrofia miotónica e acondroplasia) (Norbury & Norbury 2008; Raymond et al. 2010).
Um dos recentes avanços ao nível do cffDNA é por exemplo o seu uso para pesquisa de fetos com trissomia 21, através da técnica de sequenciação genómica paralela massiva. Esta técnica pode identificar e quantificar milhões de fragmentos de DNA de amostras biológicas em apenas poucos dias (Chiu et al. 2011). De uma maneira geral, com este procedimento pretende-se determinar a proporção de moléculas de DNA pretende-sequenciadas referentes ao cromossoma 21. Esta proporção espera-se ser elevada no plasma materno numa gravidez de um feto com trissomia 21 (Chiu et al. 2011). No entanto, é de salientar que este procedimento ainda não se encontra validado para fins de diagnóstico.
1.1.3.
Técnicas de DPN invasivas
Os procedimentos invasivos envolvem o exame direto das células ou tecidos fetais. A citogenética convencional, os métodos moleculares e bioquímicos (realizados em células cultivadas ou não cultivadas) são os usados mais frequentemente em DPN invasivo (Agnieszka
Pá
gin
a
5
et al. 2007). De entre as técnicas de DPN invasivas podem-se destacar: a biópsia do trofoblasto, a amniocentese, a cordocentese e a biópsia de tecidos fetais, as quais serão de seguida abordadas sucintamente.
1.1.3.1. Biópsia do trofoblasto
As amostras de CVS obtidas por esta técnica têm como principal vantagem a possibilidade de um diagnóstico precoce e a oportunidade de se poderem verificar posteriormente os resultados por outros métodos invasivos (Agnieszka et al. 2007). Nesta técnica, uma amostra da placenta em desenvolvimento é obtida transcervicalmente ou transabdominalmente entre a 8.ª e a 11.ª semana de gestação sob orientação ecográfica (figura 1.1).
Figura 1.1 – Amostra de células do trofoblasto obtida transabdominalmente, com auxílio de uma sonda ecográfica e de uma agulha [adaptado de (Binns & Hsu 2001)].
Vários procedimentos de diagnóstico podem ser empregues com células do trofoblasto:
1) Análise do cariótipo – deteta alterações cromossómicas numéricas e muitas alterações
cromossómicas estruturais.
2) Pesquisa de microdeleções e microduplicações por citogenética molecular.
3) Estudos enzimáticos, por exemplo, quando há um risco de erros inatos do metabolismo
(por exemplo, fenilcetonúria e doença de Gaucher).
4) Pesquisa de mutações em doenças monogénicas (Agnieszka et al. 2007).
O risco de aborto com este procedimento pode alcançar valores de 1% (Wieacker & Steinhard 2010), sendo que esta percentagem varia de acordo com a experiência do clínico. O
Pá
gin
a
6
mosaicismo confinado à placenta pode ser encontrado em 1% das amostras de CVS (Collins & Impey 2012) sendo consideravelmente maior do que para amostras obtidas por amniocentese, a qual pode ser oferecida no caso de necessidade posterior de confirmação do resultado. Além disso, outro dos problemas associados com este tipo de amostra é a contaminação pelo tecido materno (Keagle 2006).
1.1.3.2. Amniocentese
A amniocentese pode ser realizada a partir da 13.ª e 15.ª semana de gestação (amniocentese precoce) sendo usualmente feita entre a 16.ª e a 18.ª semana de gestação. A ecografia é realizada antes da amniocentese para a determinação da idade gestacional, localização da placenta, volume de fluido amniótico, atividade cardíaca fetal, número de fetos e outros fatores uterinos. Esta informação condiciona o sítio de inserção da agulha (Chodirker et al. 2001), para a recolha. A colheita é feita transabdominalmente de amostra de cerca de 1 mL de LA por semana de gestação sob orientação ecográfica (figura 1.2).
Os métodos de análise empregues incluem: o cariótipo, análises de DNA (por exemplo, diagnóstico de doenças monogénicas, tais como, hiperplasia adrenal congénita e fibrose quística) e estudos bioquímicos (por exemplo, medição dos níveis de acetilcolinesterase e α-fetoproteína quando consideramos os defeitos do tubo neural e medição da 17 α-hidroprogesterona quando há um risco de hiperplasia adrenal congénita) (Agnieszka et al. 2007).
O motivo mais comum para realizar uma amniocentese para estudos genéticos é a idade materna avançada (Keagle 2006). Estas mulheres têm um risco aumentado de terem um feto com uma aneuploidia, especialmente trissomia 21. A amniocentese é também indicada quando um progenitor é conhecido como sendo portador de um rearranjo cromossómico equilibrado, que poderá passar à descendência numa forma desequilibrada. Este procedimento é também realizado após a identificação de anomalias fetais na ecografia e para clarificar possíveis mosaicismos observados em amostras de CVS (Keagle 2006). Contudo, o risco da amniocentese associado a aborto, perda momentânea de LA e infeção intrauterina ronda os 0,5 a 1% (Wieacker & Steinhard 2010) e depende da experiência do clínico.
Pá
gin
a
7
Figura 1.2 – DPN por amniocentese. Com auxílio de uma sonda ecográfica e de uma agulha é retirado LA, a partir do qual podem ser efetuadas inúmeras análises, por exemplo, estudos de DNA [adaptado de (Binns & Hsu 2001)].
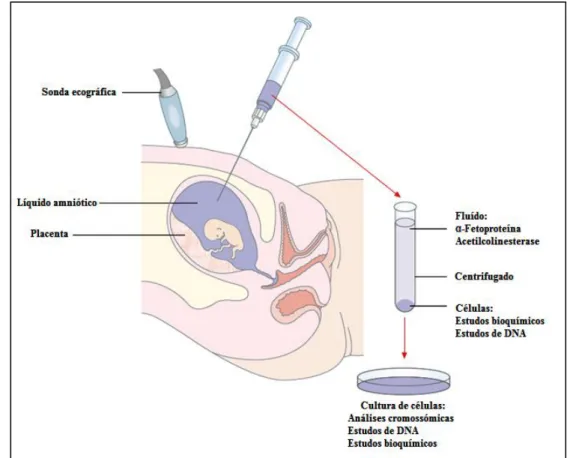
1.1.3.3. Cordocentese
A maior vantagem da cordocentese é que esta permite o acesso direto ao feto, não apenas para diagnóstico mas também para gestão terapêutica (Chodirker et al. 2001). Este procedimento pode ser usado em terapia fetal quando são requeridas transfusões intravasculares, bem como para a introdução de medicamentos em tratamento fetal.
Nesta técnica, uma amostra de 1 a 3 mL de sangue fetal é obtida a partir da veia umbilical, usualmente entre a 18.ª e a 23.ª semana de gestação sob orientação ecográfica (figura 1.3).
A amostra de sangue fetal pode ser usada para estudos genéticos e bioquímicos, incluindo análise cromossómica e diagnóstico de doenças monogénicas (por exemplo, fibrose quística). Além disso, é também possível detetar hemoglobinopatias, síndromes de deficiência imunológica e infeções intrauterinas (por exemplo, toxoplasmose) (Agnieszka et al. 2007). As indicações para a realização de cordocentese incluem: malformações congénitas ou atraso no crescimento intrauterino identificadas por ecografia (Wieacker & Steinhard 2010); infeções
Pá
gin
a
8
virais; anomalias hematológicas incluindo Rh ou outras doenças hemolíticas imunes; distúrbios ao nível das plaquetas e erros inatos do metabolismo (Chodirker et al. 2001).
Figura 1.3 – Cordocentese em DPN. Com o auxílio de uma sonda ecográfica e de uma agulha é retirado sangue fetal a partir do cordão umbilical [adaptado de (Binns & Hsu 2001)].
O risco desta técnica é estimado à volta dos 2-5%, o qual varia de acordo com a experiência do clínico, estando as complicações mais frequentes associadas com morte fetal, parto prematuro, hemorragia e bradicardia fetal (usualmente de curta duração) (Agnieszka et al. 2007).
1.1.3.4. Biópsia de tecidos fetais
A biópsia de pele realiza-se entre a 18.ª e a 20.ª semana de gestação (Fonseca et al. 2000) quando, por exemplo é necessária uma amostra não hematológica, especialmente em casos de suspeita de doenças do foro dermatológico. As biópsias de pele são muito raramente realizadas durante a gravidez em fetos em desenvolvimento. Este procedimento tem como principais desvantagens o risco de infeção, de hemorragia ou de parto prematuro, que se traduzem num risco de aborto de 5% (Fonseca et al. 2000). No entanto, este método já é mais frequente no caso dos nados-mortos após expulsão, em que amostras de tecidos sólidos podem ser usadas para detetar alterações cromossómicas quando o sangue não está disponível e os produtos de conceção são úteis na determinação da causa de muitos abortos espontâneos (Keagle 2006).
1.1.4.
Diagnóstico pós-natal
No caso do diagnóstico pós-natal, o tipo de amostra mais utilizada é o sangue periférico, podendo ser solicitado para estudos de cariótipo constitucional, estudos de MLPA e de aCGH
Pá
gin
a
9
(array Comparative Genomic Hybridization). No entanto, em determinadas situações pode-se recorrer também a amostras de fibroblastos, como por exemplo, no caso dos fetos malformados ou nados-mortos de etiologia desconhecida.
Em diagnóstico pós-natal, as indicações clínicas principais para o estudo do cariótipo complementado ou não com outras tecnologias, incluem:
Fenótipo clínico anormal ou dismorfismos; Anomalias congénitas múltiplas;
Atraso mental ou atraso do desenvolvimento psicomotor; Suspeita de síndrome de deleção /duplicação;
Doenças associadas ao cromossoma X;
Feto expulso malformado ou nado-morto de etiologia desconhecida; Presença de uma história familiar significativa de rearranjo cromossómico; Paciente com amenorreia primária ou secundária ou menopausa precoce; Azoospermia ou oligospermia severa;
Alteração clinicamente significativa do crescimento: baixa estatura, crescimento excessivo, microcefalia, macrocefalia;
Ambiguidade sexual;
Casais com deteção pré-natal de alteração cromossómica ou variante invulgar; Filho com alteração cromossómica;
Infertilidade de etiologia desconhecida; Três ou mais abortos consecutivos.
De entre todas as indicações para diagnóstico pós-natal, o atraso mental é a que representa maior impacto ao nível socioecónomico, para além de estar presente como uma consequência fenotípica principal na maioria das síndromes/distúrbios genéticos em análise neste trabalho. Por este motivo este distúrbio será abordado numa perspetiva geral, salientando alguns dos tipos de alterações cromossómicas a ele associadas (alterações subteloméricas, microdeleções/microduplicações).
O atraso mental é definido como uma diminuição significativa nas funções cognitivas [quociente de inteligência (QI) inferior a 70] e adaptativas sociais, com início antes dos 18 anos de idade (Wu et al. 2010; Rafati et al. 2012). Baseado no score do QI, o atraso mental pode ser classificado como “borderline”, ligeiro, moderado, severo e profundo (Ferrari 2009). O atraso mental como uma manifestação variável e heterogénea de disfunção do sistema nervoso central (Thuresson et al. 2007) ocorre em 2 a 3% da população, sendo que na maioria dos casos a sua
Pá
gin
a
10
causa etiológica é desconhecida (de Vries et al. 2005). O diagnóstico etiológico é um desafio, porque diversos fatores genéticos e ambientais podem contribuir para a sua patogénese (Ropers 2008). Acredita-se que os fatores genéticos estão envolvidos em muitos dos casos não diagnosticados, uma vez que há geralmente um aumento do risco de recorrência em irmãos (Crow & Tolmie 1998) e um grande número de mutações genéticas diferentes são conhecidas por estarem associadas com o atraso mental. Estima-se que 25 a 50% do atraso no desenvolvimento e atraso mental (moderado a profundo) é de etiologia genética (Wu et al. 2010) sendo que, atualmente, cerca de 282 genes humanos são conhecidos como causadores deste distúrbio (Chaudhary 2011). A síndrome do X frágil é a causa mais comum de atraso mental hereditário com uma incidência de aproximadamente 1 em cada 4000 homens e 1 em cada 7000 mulheres (Rejeb et al. 2011). Mutações em genes ligados ao cromossoma X são úteis para explicar a razão pela qual um maior número de homens, comparativamente ao número de mulheres, são severamente afetados pelo atraso mental (Chaudhary 2011).
Para além da síndrome do X frágil, a trissomia do cromossoma 21, os rearranjos subteloméricos e as microdeleções/microduplicações, representam uma fração significativa das alterações cromossómicas conhecidas associadas ao atraso mental (Rooms et al. 2004). As regiões subteloméricas são normalmente ricas em genes, e são mais suscetíveis para rearranjos do que outras regiões cromossómicas (Wu et al. 2010). Embora, os desequilíbrios subteloméricos estejam envolvidos no atraso no desenvolvimento, atraso mental ou em anomalias congénitas múltiplas, a relação causa e efeito não se apresenta bem definida (Shaw-Smith et al. 2004; Di Bella et al. 2006; Roos et al. 2006; Wu et al. 2010). As consequências clínicas são provavelmente determinadas pela localização e tipo de rearranjo, tais como deleções ou duplicações, bem como o tamanho das alterações, incluindo o número e função dos genes envolvidos (Wu et al. 2010). Essas regiões alteradas provavelmente contêm genes candidatos ainda não descritos que estão associados com o atraso no desenvolvimento/atraso mental (Wu et al. 2010). Algumas deleções submicroscópicas nas regiões subteloméricas resultam em síndromes de atraso mental bem definidas, tais como a monossomia 1p36, a síndrome de Wolf-Hirschhorn (4p-) e a síndrome cri-du-chat (5p-), mas para a maioria das alterações subteloméricas não existe um fenótipo característico associado. Por este motivo o screening de todos os subteloméros representa uma ferramenta de diagnóstico valiosa (Koolen et al. 2004).
As síndromes de microdeleção/microduplicação (quadro 1.1) são conhecidas por estarem associadas ao atraso mental, a malformações (Kjaergaard et al. 2010) e a outras anomalias, tais como, defeitos cardíacos (Shuib et al. 2006). As síndromes de microdeleção, são síndromes genéticas associadas a pequenas deleções cromossómicas, as quais usualmente envolvem 1 a 3
Pá
gin
a
11
milhões de pares de bases de DNA (Shuib et al. 2006). A variação nas manifestações fenotípicas está relacionada com a quantidade de material genético perdido na deleção cromossómica.
Quadro 1.1 – Síndromes de microdeleção/microduplicação e gene/locus envolvido (s) em cada uma delas, com a respetiva localização cromossómica [adaptado de (Theisen & Shaffer 2010)].
Os fenótipos clínicos de muitas síndromes de microdeleção definidas são variáveis, (Sharp et al. 2008; Buysse et al. 2009), enfatizando o papel de outros fatores genéticos e ambientais na determinação final do fenótipo. Algumas das síndromes de microdeleção mais comuns incluem as síndromes de Prader-Willi, Angelman, Williams, DiGeorge, Miller-Dieker, Kallmann e a síndrome de Smith-Magenis (Shuib et al. 2006). Para a maioria das síndromes de
Síndrome de microdeleção/microduplicação Gene/locus Localização
Microdeleção 1p36 Múltiplos 1p36
Microdeleção 1q21.1 com suscetibilidade para atraso mental, autismo, ou anomalias congénitas
Múltiplos 1q21.1 Microdeleção 3q29 Múltiplos 3q29 Microduplicação 15q11-15q13 Múltiplos 15q11-15q13 Microdeleção 15q13.3 Múltiplos 15q13.3 Microdeleção 16p11.2p12.2 Múltiplos 16p11.2p12.2 Microdeleção 17q21.31 Múltiplos 17q21.31
Microdeleção distal 22q11.2 Múltiplos 22q11.2
Microduplicação 22q11.21 Múltiplos 22q11.21
Angelman UBE3A 15q11.2
Beckwith-Wiedemann, relacionado IGF2 IGF2 11p15.5
Cat eye Múltiplos 22q11.1
Charcot-Marie-Tooth Tipo 1A PMP22 17p12
Cri du chat Múltiplos 5p15.2
DiGeorge I/velocardiofacial HIRA TBX1 22q11.21
DiGeorge 2 Múltiplos 10p14
Distrofia muscular Duchenne DMD Xp21.2-p21.1
Kallmann I KALI Xp22.31 Langer–Giedion TRPS1 EXT1 8q23.3 8q24.11 Miller–Dieker/Lisencefalia 1 PAFAH1B1 (LIS1) 17p13.3
Microduplicação Potocki–Lupski/17p11.2 Múltiplos 17p11.2
Prader–Willi SNRPN 15q11.2 Retinoblastoma/MR RB1 13q14.2 Rubinstein–Taybi CREBBP 16p13.3 Smith–Magenis RAI1 17p11.2 Williams–Beuren ELN 7q11.23 Wolf–Hirschhorn Múltiplos 4p16.3
![Figura 1.1 – Amostra de células do trofoblasto obtida transabdominalmente, com auxílio de uma sonda ecográfica e de uma agulha [adaptado de (Binns & Hsu 2001)]](https://thumb-eu.123doks.com/thumbv2/123dok_br/15952656.1097766/31.892.181.734.414.773/figura-amostra-células-trofoblasto-transabdominalmente-auxílio-ecográfica-adaptado.webp)
![Figura 1.3 – Cordocentese em DPN. Com o auxílio de uma sonda ecográfica e de uma agulha é retirado sangue fetal a partir do cordão umbilical [adaptado de (Binns & Hsu 2001)]](https://thumb-eu.123doks.com/thumbv2/123dok_br/15952656.1097766/34.892.124.774.158.486/figura-cordocentese-auxílio-ecográfica-retirado-cordão-umbilical-adaptado.webp)