U
NIVERSIDADE DE
L
ISBOA
F
ACULDADE DE
C
IÊNCIAS
D
EPARTAMENTO DE
B
IOLOGIA
V
EGETAL
R
EGULATION OF THE MECHANISM OF
INTERFERENCE WITH
Q
UORUM
SENSING IN
E
SCHERICHIA COLI
Paulo José Braz Correia
M
ESTRADO EM
M
ICROBIOLOGIA
A
PLICADA
U
NIVERSIDADE DE
L
ISBOA
F
ACULDADE DE
C
IÊNCIAS
D
EPARTAMENTO DE
B
IOLOGIA
V
EGETAL
R
EGULATION OF THE MECHANISM OF
INTERFERENCE WITH
Q
UORUM
SENSING IN
E
SCHERICHIA COLI
Dissertação orientada por:
Doutora Karina Xavier (IGC) e Professor Doutor Mário Santos (FCUL)
Paulo José Braz Correia
M
ESTRADO EM
M
ICROBIOLOGIA
A
PLICADA
R
EGULATION OF THE MECHANISM OF
INTERFERENCE WITH
Q
UORUM
SENSING IN
E
SCHERICHIA COLI
Paulo José Braz Correia
M
ASTER
T
HESIS
2011
This thesis was fully performed at the Bacterial Signaling Laboratory in
the Instituto Gulbenkian de Ciência under the direct supervision of
Dr. Karina Xavier.
Professor Mário Santos was the internal designated supervisor in the
scope of the Master in Applied Microbiology of the Faculty of Sciences of
the University of Lisbon.
i
Acknowledgements
First and foremost I offer my sincerest gratitude to my supervisor, Dr. Karina Xavier, who has supported me throughout my thesis with her knowledge and expertise. I could attribute an important part of my Masters degree to her encouragement and guidance. One simply could not wish for a better or friendlier supervisor.
I would also like to thank to Professor Mário Santos, my internal supervisor, for the support and advice.
I would like to thank all the members of the group, the bacterial signaling group that received me well in the group and helped me a lot during this journey. To Catarina, for all the support, for the important advice and for helping me in conquering the PTS; To Rita, for helping me in laboratory and for the profitable discussions about our work; to João, for his knowledge and for his devotion to science and to Jess and Pol, for what they taught me, for all their experience in science and for the endless laughs.
I would also like to thank my family:
A toda a minha família, em especial aos meus avós, pelo pensamento sempre presente e por terem contribuído para o que sou e o que alcancei nesta vida.
Aos meus irmãos: ao Ricardo “Mourinho” pela boa disposição, confiança e pelos jantarinhos que fez para mim durante o processo de concepção desta dissertação. Ao Pedro “Armstrong”, pela experiência em mestrados, pelos treinos que me orientou e pelo discurso motivacional importantíssimo nestas alturas.
Aos meus pais que fizeram deste “menino” aquilo que ele é. Ao meu pai, pela motivação e apoio que me transmite e por me permitir pensar em momentos de desmotivação: “Mas quem sou eu? Sou filho do Zé Correia, vou acabar esta tese de certeza!”; à minha mãe pelo seu discurso tranquilizador, pelas energias positivas que me passa, pelo carinho, pelo apoio e por tudo. Este “bambino” tem muito orgulho de ser vosso filho.
Ao meu amor, Joana Sousa, pela compreensão, pela motivação, pelo apoio, pelo amor, pelo suporte emocional que me deu durante esta etapa da minha vida e por ser tão especial.
ii
Abstract
Quorum sensing is a cell-to-cell signaling mechanism in which bacteria collectively control gene expression and therefore synchronize behaviors only productive at a high population density. The autoinducer-2 (AI-2) signal is produced by many bacteria and enables inter-species communication. In Escherichia coli, AI-2 is synthesized and secreted, accumulating in the extracellular medium. AI-2 concentration rises as bacteria divide and decreases rapidly in early stationary phase partially due to the expression of an ATP-binding cassette transporter, named Lsr (for LuxS-regulated) and encoded by the lsrACDB operon, which imports AI-2, removing it from the environment.
Recent work showed that the phosphoenolpyruvate-dependent phosphotransferase system (PTS) plays an important role in the regulatory network of the Lsr transporter. Specifically, mutants in the ptsI gene, encoding for Enzyme I (EI) of the PTS, do not internalize extracellular AI-2 and do not activate the lsr operon. The present work showed that phosphorylation of EI is important for enabling the recovery of the defect of the ptsI mutation in AI-2 internalization but most likely not to phosphorylate AI-2 since LsrK kinase is required for AI-2 processing.
In this study a genetic screen was performed to determine the molecular mechanism of the regulation of AI-2 internalization via EI by identifying suppressors of the ptsI mutant that can incorporate AI-2. A library of single-gene deletions of all non-essential genes in
Escherichia coli in a ptsI deletion background strain carrying a lsr-lacZ promoter fusion was
generated and screened for mutants with lsr-lacZ expression higher than the parent strain. Suppressors of the ptsI mutant were identified and the most interesting mutants are currently being analyzed to understand their role.
The Lsr regulation was also studied using a new tool: green fluorescent protein (gfp) reporter fusions. These were compared with the commonly used promoter-lacZ fusions and their benefits and drawbacks were accessed.
Understanding how the Lsr transport is dependent on the PTS, can reveal the molecular mechanism through which information about the physiological state of bacteria and regulation of AI-2 signal uptake is integrated.
iii
Resumo
A detecção de quórum é um mecanismo de sinalização entre células, no qual as bactérias colectivamente controlam a expressão de genes e desta forma sincronizam comportamentos que apenas são eficientes a uma elevada densidade populacional. Este processo é caracterizado pela produção, pela secreção e pela detecção de pequenas moléculas de sinalização denominadas autoindutores. A concentração extracelular destes indutores encontra-se directamente relacionada com o número de indivíduos na população.
Um desses indutores denomina-se autoindutor-2 (AI-2) que é um sinal produzido por muitas bactérias e constitui a primeira molécula a ser identificada que promove a comunicação célula-a-célula entre diferentes espécies bacterianas. Possibilita a comunicação inter-espécies, regulando vários fenótipos como a bioluminescência e a formação de biofilmes.
Nas bactérias Salmonella Typhimurium e Escherichia coli, o AI-2 é sintetizado e secretado, acumulando-se no meio extracelular e a sua concentração aumenta à medida que as bactérias se vão dividindo. A actividade extracelular do AI-2 é máxima durante a fase exponencial tardia do crescimento bacteriano e decresce rapidamente durante a entrada na fase estacionária. Esta rápida internalização do AI-2 deve-se à expressão de um transportador do tipo ABC (do inglês “ATP-binding cassette”), denominado sistema de transporte Lsr (de LuxS-regulated) e que é responsável pela incorporação daquele sinal, possibilitando a sua fosforilação e processamento no interior da célula bacteriana. Este transportador é codificado pelo operão lsrACDB.
Este sistema, o transportador Lsr, apresenta dois importantes reguladores: o LsrR, que é o repressor transcricional do operão lsr e a LsrK, que é a cinase que fosforila o AI-2 que é incorporado, a AI-2-fosfato (AI-2-P). Esta molécula activada liga-se ao regulador LsrR e a repressão por si mediada é removida, permitindo a transcrição do operão lsr e a formação do sistema de transporte Lsr. Estes reguladores são codificados pelos genes lsrR e lsrK, respectivamente, localizados imediatamente a montante do operão lsr e divergentemente transcritos na direcção oposta.
O modelo actual para a incorporação do AI-2 assume a premissa que a produção do transportador Lsr se encontra reprimida pelo LsrR para evitar a expressão prematura do mecanismo de incorporação do AI-2, permitindo a acumulação de AI-2 no meio extracelular. Assim, se o LsrR se encontra a reprimir o operão lsr, não existe transcrição do transportador Lsr e supostamente não há internalização de AI-2.
Contudo, na transição para a fase estacionária, a internalização do AI-2 inicia-se e para começar este processo, a fosforilação do AI-2 intracelular é necessária para inactivar a
iv repressão mediada pelo LsrR. Assim, isto só seria possível se existisse um outro transportador de baixa afinidade que fosse capaz de incorporar o AI-2. O AI-2 seria posteriormente fosforilado e iria desencadear o sistema, permitindo a indução da expressão do transportador Lsr e que iria rapidamente incorporar o AI-2 presente no meio extracelular.
Estudos recentemente efectuados demonstraram que a enzima I (EI) do Sistema das Fosfotransferases dependente do fosfoenolpiruvato (PTS) desempenha um papel importante na rede regulatória do transportador Lsr. Concretamente, foi observado que mutantes no gene ptsI, que codifica a EI, não internalizam o AI-2, nem activa a transcrição do operão lsr.
Este facto, envolvendo o PTS na incorporação do AI-2, enquadra-se perfeitamente na observação de que mutantes no transportador Lsr apresentam uma incorporação lenta do AI-2 e que esta não é abolida. Uma vez que o AI-2 não se difunde passivamente através da membrana, um transportador desconhecido é capaz de importar este sinal. De acordo com isto e com o que foi observado no mutante ptsI, é plausível que o PTS, um importante sistema envolvido na incorporação de hidratos de carbono e na sua regulação, possa estar envolvido na internalização do AI-2 ou na regulação deste processo. De facto, foi igualmente demonstrado que a incapacidade do mutante ptsI para internalizar o AI-2 se deve, efectivamente, à incapacidade do AI-2 para entrar na célula. Apesar disto, o mecanismo dependente do PTS, que permite a incorporação de AI-2, é ainda desconhecido.
Neste trabalho a importância da actividade fosfotransferase da EI para a incorporação do AI-2 e para a activação do operão lsr foi determinada. O mutante ptsI foi complementado usando um plasmídio induzido por IPTG e que continha um gene mutante do ptsI que codifica uma EI que não pode ser fosforilada (EIH189A), uma vez que se encontrava mutada no local de fosforilação e, apesar disso, a internalização de AI-2 não se verificou. Por outro lado, quando um alelo selvagem do gene ptsI é fornecido, a capacidade para incorporar AI-2 e a activação do operão lsr são repostas. Os resultados indicam que uma EI funcional, com um local de fosforilação preservado, é necessário para a incorporação de AI-2 e para a transcrição do operão lsr.
Assim, a internalização do AI-2 podia ser efectuada por uma das 20 permeases PTS, específicas para cada hidrato de carbono ou por uma permease que não pertence à família PTS mas que é regulada por componentes do PTS, onde a EI e a sua actividade de fosfotransferase desempenharia um papel crucial neste processo.
Neste estudo foi efectuado um rastreio genético para determinar o mecanismo molecular envolvido na regulação da internalização de AI-2 via EI, através da identificação de supressores da mutação ptsI, capazes de incorporar AI-2. Para isto, foi gerada uma biblioteca de delecções únicas de todos os genes não essenciais de Escherichia coli, num mutante no gene ptsI, que possuía uma fusão lsr-lacZ e procuraram-se mutantes duplos com uma expressão da fusão lsr-lacZ maior do que a estirpe parental. Este rastreio genético
v provou ser bem sucedido na identificação de supressores do mutante ptsI e neste momento os mutantes mais interessantes estão as ser estudados para compreender o seu papel na interacção entre o Sistema das Fosfotransferases e o sistema Lsr.
Os genes que foram definidos como candidatos positivos no rastreio genético podem ser classificados em várias classes: genes codificando componentes do PTS ou reguladores do PTS; genes que codificam transportadores não PTS, como simportes, antiportes e transportadores ABC; genes codificando reguladores de transcrição que se ligam ao DNA; metiltransferases; genes com função desconhecida e genes com outras funções.
Para além do rastreio genético, neste trabalho de investigação, outro instrumento foi implementado para o estudo da regulação do operão lsr, em vez das fusões com o gene
lacZ, normalmente usadas, e dos ensaios de actividade da enzima β-galactosidase. Diferentes fusões repórter com o gene da proteína verde fluorescente foram construídas para avaliar a expressão do operão em diferentes estirpes e para as comparar com as fusões anteriores. O objectivo principal foi a implementação de novos métodos de estudo do operão lsr e a sua regulação em Escherichia coli.
Em conjunto, os resultados obtidos neste trabalho de investigação mostram que o gene
lsrK é transcrito juntamente com o gene lsrR pelo promotor lsrR e encontra-se sujeito à sua
regulação. Como este promotor é mais forte do que o promotor do operão lsr, é menos reprimido pelo LsrR e transcrito a baixos níveis, mesmo na ausência do AI-2. Esta transcrição basal do promotor lsrR e desta forma dos genes lsrR e lsrK, antes da expressão do sistema de transporte Lsr, possibilita a presença do repressor da transcrição para evitar a indução prematura do sistema; e permite que a cinase LsrK fosforile o AI-2 que é primeiro internalizado pelo transporte dependente do sistema PTS, produzindo AI-2-P que se liga ao repressor LsrR, removendo a repressão por si mediada e promovendo a indução da expressão do transportador Lsr que rapidamente incorporará o AI-2 do meio extracellular.
A compreensão do mecanismo de como o sistema de transporte Lsr se encontra dependente do PTS, pode revelar o mecanismo molecular através do qual a informação referente ao estado fisiológico das bactérias e a regulação da incorporação do AI-2 podem ser integradas.
Contents
Acknowledgements ...i
Abstract ... ii
Resumo ... iii
1. Introduction ...1
1.1. Quorum sensing: bacterial communication...1
1.2. Autoinducer-2 signal molecule: inter-species quorum sensing ...3
1.3. AI-2-mediated regulation in Escherichia coli and Salmonella enterica Typhimurium...6
1.4. The Phosphoenolpyruvate-dependent Phosphotransferase System (PTS) ...9
2. Materials and Methods ... 12
2.1. Bacterial strains and growth conditions ... 12
2.2. β-galactosidase assays ... 12
2.3. AI-2 activity assay ... 12
2.4. Plasmid construction ... 13
2.5. DNA manipulations ... 13
2.6. Green fluorescent protein expression assay ... 13
2.7. Construction of the strains carrying the lsrR-lacZ promoter fusion ... 14
2.8. Screen for regulators of EI of the PTS and suppressors of the ptsI mutation ... 14
2.8.1. Preparation of P1 bacteriophage lysates in 96-well plates... 15
2.8.2. P1 transduction in 96-well plates ... 15
2.8.3. Screening using MacConkey Lactose Agar plates ... 15
3. Results ... 18
3.1. The role of EI of PTS in AI-2 internalization ... 18
3.1.1. A functional EI is required for AI-2 incorporation and lsr operon expression ... 18
3.1.2. Mlc, an important regulator of PTS, does not play a role in Lsr regulation ... 21
3.1.3. Screen for genes that suppress inhibition of AI-2 internalization in a ptsI mutant ... 22
3.2. Studying regulation of the lsr operon using transcriptional fusions ... 29
3.2.1. Studying regulation of lsrACDBFG operon using a gfp reporter fusion ... 29
3.2.2 Studying regulation of the lsrRK operon using gfp reporter fusions ... 31
3.2.3. Comparing the regulation of the lsrR and lsr promoters ... 34
3.2.4. Regulation of the lsrK gene expression ... 35
4. Discussion ... 37
1
1. Introduction
1.1. Quorum sensing: bacterial communication
The conventional view of bacterial existence always considered bacteria as independent individuals, living their sole lives and capable of reacting to changes in the environment but not communicating directly with other microorganisms in the vicinity. This view persisted for long but currently it is well established that bacteria have elaborated chemical signaling systems that enable them to communicate within and between species through a mechanism designated quorum sensing [1-3].
Quorum sensing is a cell-to-cell signaling mechanism in which bacteria collectively control gene expression and therefore synchronize behaviors that are only productive at a high population density. This process is characterized by the production, secretion and detection of small signaling molecules named autoinducers. The extracellular concentration of the autoinducers correlates directly with the number of individuals in the population. Thus, when the autoinducer reaches a certain concentration in the extracelular medium, corresponding to a quorum of bacteria, a signal cascade is triggered resulting in a coordinated modification of the gene expression of the population allowing the bacteria to carry out behaviors collectively or as a group. Some of these bacterial processes are formation of biofilms, secretion of virulence factors, production of toxins, bioluminescence, competence for DNA uptake and sporulation [1, 4, 5].
Most quorum sensing communication systems can be organized into two major classes that differ according to the type of signaling molecules: the LuxI/LuxR-type systems using the acyl-homoserine lactones (AHL) in the Gram-negative bacteria and the two-component signaling circuits based on modified oligopeptides that are characteristic of the Gram-positive bacteria. Both quorum sensing systems can be characterized by their high specificity and function for intraspecies communication [1, 6]
The AHL autoinducer signals typically are species specific signals that enable intraspecies cell-to-cell communication [7, 8]. These molecules are synthesized by the LuxI-type enzymes which promote the ligation of an acyl moiety from a fatty acyl carrier protein, to the lactonized methionine moiety of S-adenosylmethionine (SAM) [9, 10]. The AHL can present different forms, but they are characterized by their common core, the homoserine lactone ring, which carries distinct acyl side chains of C4 to C18 in length conferring specificity to the AHL autoinducer [11]. The majority of the AHL crosses the cellular membranes freely by diffusion, due to their size and polarity, but few AHL need to be actively transported [7]. As cells proliferate AHL concentration increase and when it reaches a threshold concentration it is detected in the cytoplasm by a LuxR-type protein, the response regulator, to which it binds. These LuxR-AHL complexes recognize and bind specific consensus sequences of DNA
2 promoter elements and activate the transcription of quorum sensing controlled genes (Figure 1A) [1, 11]. This communication system, comprising the AHLs and the LuxI/LuxR signaling mechanism, was initially discovered in the marine bacterium Vibrio fischeri where it controls the expression of the luciferase operon and, therefore the production of bioluminescence [12-14]. This LuxI/LuxR signaling mechanism is considered the paradigm for the control of gene expression by quorum sensing in the Gram-negative bacteria and regulates virulence in human pathogens such as Pseudomonas aeruginosa [15] and Vibrio cholerae [16].
In the Gram-positive bacteria, as stated above, the distinctive autoinducers are modified oligopeptides that control the expression of the quorum sensing genes [17-19]. These autoinducing peptides (AIPs), encoded in the bacterial genome, consist of 5-17 amino acids and are highly species or even strain specific [20].
The AIPs are initially synthesized as large precursor peptides that undergo post-translational modifications such as cyclization and processing, including the incorporation of lactone and thiolactone rings, lanthionines and isoprenyl groups [21-25]. After these modifications or in a concomitant manner, the AIPs are actively secreted out of the cell by specialized transporters since the bacterial membranes are impermeable to them, not allowing their simple diffusion. Once outside the cell, the concentration of the AIPs increases as the cell density increases and the AIPs are recognized by external domains of membrane-bound sensor histidine kinase which is part of a two-component system (Figure 1B).
Figure 1 – The two major species specific quorum sensing systems. A) Canonical Gram-negative
LuxI/LuxR quorum sensing system. Red pentagons represent the acyl-homoserine lactones. LuxI is the autoinducer synthase and LuxR is the response regulator that binds AHL and activates the transcription of quorum sensing genes B) Model for Gram-positive oligopeptides/two-component system quorum sensing mechanism. Blue octagons are the autoinducing peptides after processing or modification. The histidine kinase (H) undergoes autophosphorylation when the autoinducing peptide binds. The phosphate is then transferred to a response regulator (D) that changes its DNA binding activity and regulates the expression of quorum sensing target genes (figure from reference [1]).
3 The sensor histidine kinase undergoes autophosphorylation when the AIP binds and interacts with the response regulator, phosphorylating it. The activated response regulator, the second protein of the two-component system, regulates the expression of quorum sensing target genes [1, 6, 26]. This peptide signaling system plays a role in important bacterial processes by regulating the development of bacterial competence in Bacillus
subtilis and Streptococcus pneumoniae, conjugation in Enterococcus faecalis and virulence
in Staphylococcus aureus [27, 28].
Although these mechanisms of bacterial communication used by Gram-negative and Gram-positive bacteria are distinct considering the nature of the signaling molecules and the proteins involved; they have similarities since both allow microorganisms to perceive and monitor the environment, to coordinate their communities and behaviors as multicellular organisms, to face the environmental challenges and to better adapt and grow in a certain habitat. Furthermore, both foster the intra-species quorum sensing communication due to the extreme specificity of the signaling molecules [1, 6, 29, 30].
However, beyond these autoinducers, that promote communication within each species; there is one, which is so far the only signaling molecule identified that is shared by both Gram-negative and Gram-positive and produced by a wide range of bacteria: autoinducer-2 (AI-2) [31, 32].
1.2. Autoinducer-2 signal molecule: inter-species quorum sensing
Autoinducer-2 (AI-2) is a low-weight signal molecule that is synthesized and recognized by a wide variety of bacteria and constitutes the first identified molecule promoting cell-to-cell communication between different bacterial species [6, 33-35]. AI-2 was first identified in the marine bacterium Vibrio harveyi as part of a complex quorum sensing system responsible for the regulation of bioluminescence [36]. To date, AI-2 or its synthase LuxS have already been shown to regulate other important bacterial behaviors such as the formation of biofilms [37, 38] and the production of virulence factors [39, 40]. Moreover, the gene encoding the synthase, luxS, has been identified in approximately half of the bacterial sequenced genomes, supporting the idea that many bacteria produce and use AI-2 to communicate with other bacterial species [31, 34].
The biosynthetic pathway of AI-2 is conserved amongst the bacteria that produce this molecule and occurs through three enzymatic steps (Figure 2). AI-2 is synthesized by the LuxS enzyme from S-adenosylmethionine (SAM). SAM is an important methyl donor used by methyltransferases in vital cellular processes such as nucleic acid and protein methylation, in a metabolic pathway know as the activated methyl cycle [41, 42]. During the transmethylation reactions, SAM donates a methyl group being converted into the toxic metabolic intermediate
4 S-adenosylhomocysteine (SAH) and this nocuous intermediate must be eliminated by the bacterial cell. SAH is hydrolyzed to S-ribosylhomocysteine (SRH) and adenine by the nucleosidade enzyme Pfs. SRH is then cleaved to 4,5-dihydroxy-2,3-pentanedione (DPD) and homocysteine by the LuxS enzyme [35, 41, 43]. In bacteria that do not possess LuxS or in eukaryotes and archaea, SAH still needs to be detoxified. In these cases, it is hydrolysed to adenosine and homocysteine in a reaction performed by SAH hydrolase, and DPD is not produced [4, 41]. In both cases, the reactions yield homocysteine that then enters the activated methyl cycle, restoring SAM.
Figure 2 – Three-step production of DPD from SAM. SAM donates a methyl group in
transmethylation reactions performed by methyltransferases, leading to the formation of the toxic intermediate SAH. SAH is then converted to SRH and adenine by the Pfs enzyme and SRH is finally converted to DPD and homocysteine by the LuxS enzyme (figure from reference [44]).
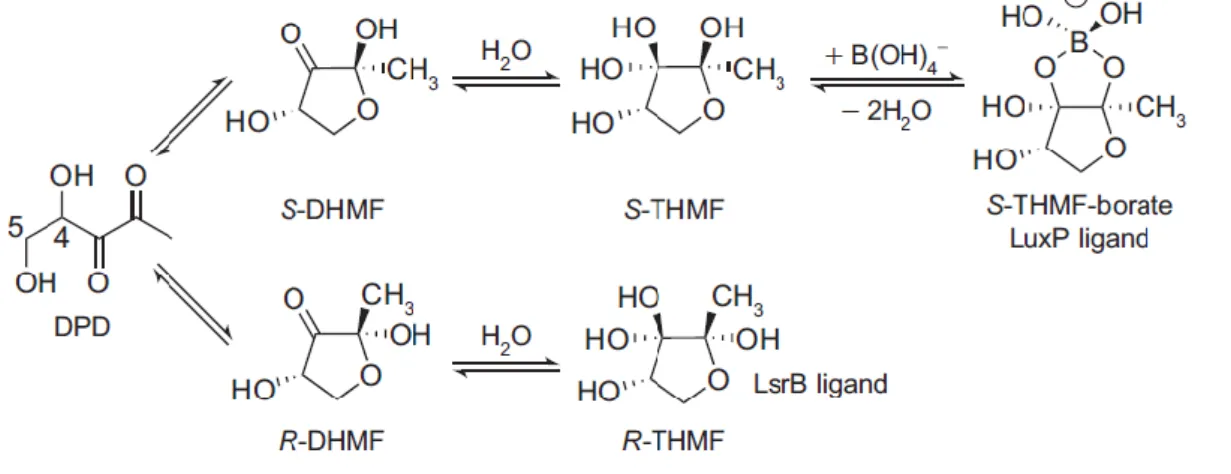
DPD is a very unstable and reactive molecule that cyclizes and, in solution, undergoes a variety of spontaneous rearrangements giving rise to a group of chemical distinct molecular forms or derivatives that exist in equilibrium [35, 43]. The set of different molecules resulting from the precursor DPD are generically designated AI-2 [34, 41]. This distinctly rearranged DPD forms are recognized by different microorganisms [35, 44].
So far, two different AI-2 conformations derived from DPD were identified (Figure 3) by trapping the active molecules in their respective receptors. Vibrio harveyi recognizes an AI-2 conformation that requires an atom of boron to be bound to the rearranged DPD derivative to be active. This form of AI-2 is a furanosyl borate diester which is called (2S,4S)-2-methyl-2,3,3,4-tetrahydroxytetrahydrofuran-borate (S-THMF-borate) and it is recognized by the LuxP receptor. In Salmonella Typhimurium a different AI-2 conformation, that does not require boron, is recognized. It is designated (2R,4S)-2-methyl-2,3,3,4-tetrahydroxytetrahydrofuran (R-THMF), presents a distinct stereochemistry and binds to the LsrB periplasmic receptor [35, 44]. Recently, it was found that this last AI-2 form is also recognized by Sinorhizobium
5 Although the AI-2 forms, which are recognized by each of those receptors, present a distinct conformation, they can interconvert spontaneously, in solution, enabling different bacterial species to respond to their own and also to AI-2 produced by others [35, 44, 46]. Nevertheless, this interconversion and the availability of each form are highly dependent on the environment and on the presence of borate, in case of the AI-2 recognized by Vibrio
harveyi [44].
Figure 3 – AI-2 is a family of interconverting molecules derived from DPD. In solution, DPD
undergoes spontaneous rearrangements and assumes the conformation of two different epimeric furanoses: R-DHMF and S-DHMF [(2R,4S)- and (2S,4S)-2,4-dihydroxy-2-methyldihydrofuran-3-one, respectively]. Hydration of R-DHMF and S-DHMF gives rise to R-THMF and S-THMF, respectively. In the presence of borate, S-THMF yields S-THMF-borate which is the ligand of LuxP receptor from
Vibrio harveyi. R-THMF, that does not bind boron is recognized by LsrB from Salmonella Typhimurium
(figure from reference [47]).
Despite the diversity of behaviors regulated by AI-2 [38], the mechanisms and the molecular details of AI-2 detection and signal transduction have only been determined in few bacteria such as Vibrio harveyi [48] and Vibrio cholerae and in the bacteria Salmonella Typhimurium, Escherichia coli [47, 49, 50] and Sinorhizobium meliloti [45].
In Salmonella Typhimurium, Escherichia coli and Sinorhizobium meliloti, AI-2 plays a regulatory role by inducing the lsr operon (from luxS-regulated) that consists of the genes responsible for encoding the components of the apparatus used for the import and processing of AI-2. This transport system is so effective that AI-2 is almost completely depleted from the extracellular medium [51].
This striking behavior led to the current idea that it represents a mechanism of interference with AI-2-regulated quorum sensing, corroborating the function of AI-2 as an universal language enabling inter-species bacterial communication [6, 35, 44, 46].
6
1.3. AI-2-mediated regulation in Escherichia coli and Salmonella enterica Typhimurium
In the enteric bacteria, Salmonella Typhimurium and Escherichia coli, as mentioned, AI-2 is synthesized and secreted, accumulating in the extracellular medium and its concentration rises as bacteria divide. AI-2 extracellular activity peaks during the mid to late exponential phase and rapidly decreases during entry into the stationary phase. This fast internalization of AI-2 is due to the expression of an ATP-binding cassette (ABC) transporter (designated Lsr from luxS-regulated) that carries out the uptake of this signal allowing its further phosphorylation and processing inside the bacterial cell [49-52].
The Lsr transport system is constituted by proteins encoded by the genes lsrACDBFG which constitute the lsr operon. This operon is regulated by cyclic AMP (cAMP) and by cAMP receptor protein (CRP) and also by two proteins transcribed from lsrK and lsrR genes, located immediately upstream of the lsr operon and that are divergently transcribed in the opposite direction [52-54]. This observation came from the fact that in lsrK mutants, the Lsr transporter expression is repressed and AI-2 remains in the extracellular milieu. On the other hand, in lsrR mutants the Lsr transporter is constitutively expressed and the extracellular AI-2 is continuously imported into the cell [52]. LsrR is a transcriptional regulator which, in the absence of AI-2, represses the lsr operon and AI-2 internalization. When extracellular concentration of AI-2 increases, it is phosphorylated to AI-2-phosphate (AI-2-P) by the cytoplasmic kinase, LsrK. This activated molecule binds LsrR, enabling the expression of the
lsr operon [50].
Specifically, the mechanism of AI-2 internalization (Figure 4) starts when AI-2 is first recognized by the periplasmic protein, LsrB, and then imported through the two transmembrane domains, LsrC and LsrD, into the bacterial cell. This transport is driven by the energy provided by ATP hydrolysis catalyzed by the ATPase, LsrA. Upon internalization, AI-2 is phosphorylated to AI-2-P by LsrK and sequestered in the cytoplasm where it binds the transcriptional repressor, LsrR, inhibiting it and avoiding its binding to the lsrACDBFG promoter, relieving the LsrR-mediated repression. As a consequence, the expression of the genes encoding the Lsr transporter is induced and upregulated. The Lsr apparatus is assembled and rapidly incorporates AI-2 from the extracellular milieu, causing a positive feedback loop, responsible for AI-2 depletion from the medium before reaching the stationary phase of growth [49-51].
In the cytoplasm, AI-2-P is further processed by the enzymes LsrG isomerase and LsrF, encoded by the other genes of the lsr operon, lsrG and lsrF, respectively [47, 55, 56].
LsrG is responsible for carrying out the isomerization of AI-2-P to 3,4,4-trihydroxy-2-pentanone-5-phosphate (P-TPO), which has an unknown function [56]. The role played by LsrF in AI-2-P processing remains unclear, as well as its substrates and products but its
7 sequence resembles an aldolase enzyme [55]. Through the action of these enzymes on AI-2-P processing occurs transcription termination of lsrACDB operon and the AI-2 signaling cycle terminates [47].
Figure 4 – AI-2 incorporation and processing by the lsrACDBFG operon in enteric bacteria. AI-2
is synthesized by the LuxS enzyme and accumulates in the extracellular medium. When AI-2 reaches a certain concentration, it binds to LsrB and is incorporated by the Lsr transport apparatus, transcribed by the lsr promoter. Once inside the cell, AI-2 is phosphorylated by the kinase LsrK yielding AI-2-P. This activated molecule binds to LsrR, the transcriptional regulator, transcribed by the lsrR promoter. The repression mediated by LsrR is relieved and the expression of the Lsr transporter by the lsr promoter is upregulated, resulting in a rapid uptake of extracellular AI-2. AI-2-P is further processed by LsrG and LsrF giving rise to the product 3,4,4-trihydroxy-2-pentanone-5-phosphate (P-TPO). In this mechanism, the promoter responsible for transcription of the lsrK gene is not determined yet (dashed arrow) (modified from reference [28]).
The biologic function of this AI-2 incorporation system remains unrevealed. However, it has been shown that the Lsr transport system of Escherichia coli interfere with AI-2 controlled behaviors of other bacterial species present in the same environment [46]. Interfering with quorum sensing pathways of bacteria that produce and detect AI-2 to regulate the expression of important genes could confer an important advantage. As a matter of fact, this was already observed when Vibrio harveyi and Escherichia coli were co-cultured together [46]. It was shown that Escherichia coli uses its Lsr transporter to internalize endogenously produced AI-2 as well as AI-2 produced by Vibrio harveyi, and the levels of this molecule rapidly drop. As a consequence, the induction of AI-2-regulated genes and behaviors in Vibrio harveyi, like bioluminescence, is prevented [46].
Evidences of this role of the Lsr transport system were also observed in Sinorhizobium
8 but, surprisingly, it eliminates the signal from the environment, using its own Lsr apparatus to incorporate AI-2 [45]. In short, the Lsr transport system can be a mechanism of interfering with AI-2-based quorum sensing processes of other species.
This model of AI-2 incorporation and processing, describes in an elegant manner the mechanism of regulation mediated by AI-2 in enteric bacteria. However, there are some questions that still remain to be addressed. According to this model of AI-2 internalization, the transcription repressor LsrR must be repressing the lsr operon to avoid premature induction of the expression of the transporter; otherwise the signal would not accumulate in the environment. As a result, if LsrR is repressing the operon, there is no transcription of the Lsr transporter and supposedly no AI-2 incorporation. Nevertheless, to start this process, it is required that AI-2 is internalized and phosphorylated to derepress LsrR. This is possible if another transporter of lower affinity is capable of importing AI-2, if so then uptake by this other transporter would trigger the whole system. This is supported by the observation that mutants defective for the Lsr transport system still incorporate AI-2, but at a lower rate [28].
In addition, in order to phosphorylate the AI-2 that is first internalized by the secondary transporter, there must be a basal level of LsrK to produce AI-2-P that induces the system. Previous work revealed that in a lsrK null background, the internalization of AI-2 is abolished and there is no activation of the Lsr system. This means that no matter which transporter is internalizing AI-2, LsrK is required to phosphorylate AI-2 and to sequester the signal inside the cell. AI-P-2 produced induces the transcription of the Lsr apparatus, by relieving LsrR-mediated repression [28].
Therefore, lsrK transcription and LsrK protein levels or activity must be somehow controlled by a different regulatory mechanism than that of the Lsr transport component. One possibility is that lsrK has its own, but weak, promoter, not dependent on LsrR neither on AI-2-P, to ensure that the kinase is always present at low-levels to trigger the system once AI-2 is imported.
In order to address the questions regarding this process, recent work from our laboratory has shown that the phosphoenolpyruvate-dependent phosphotransferase system (PTS) plays an important role in the regulatory network of the Lsr transporter. A genetic screen performed in Escherichia coli, revealed that mutants in the ptsI gene, encoding for enzyme I (EI) of the PTS, do not internalize the extracellular AI-2 as well as do not activate the lsr operon [28]. Thus, a new model for AI-2 incorporation and Lsr transporter activation was proposed. Accordingly, it is now hypothesized that uptake of extracellular AI-2 by a permease regulated by PTS is necessary to start AI-2 internalization and, that together with expression of LsrK at a basal level enable production of AI-2-P, which in turn promotes the relief of lsr operon repression and the expression of the Lsr transport system. [28]. In other words, EI and the PTS, that are responsible for the uptake of a wide range of carbohydrates
9 to the cell, are certainly involved in the first AI-2 import, through an unknown low-affinity permease. This permease is regulated by the PTS. However, most likely it is not a permease from the PTS family, because single mutants on PTS permeases do not show impairment on AI-2 incorporation comparable to EI mutants.
1.4. The Phosphoenolpyruvate-dependent Phosphotransferase System (PTS)
The phosphoenolpyruvate-dependent phosphotransferase system (PTS) is a mechanism for the translocation through the bacterial cell membrane of numerous carbohydrates, like monosaccharides, disaccharides, amino sugars, polyols and other sugar derivatives. This transport is performed with concomitant phosphorylation and uses phosphoenolpyruvate (PEP) as an energy source and phosphoryl donor [57, 58].
The PTS consists of two general cytoplasmic components, the Enzyme I (EI, encoded by the ptsI gene) and the Histidine-containing protein (HPr, encoded by the ptsH gene), which are common to all PTS carbohydrates. The specificity of each PTS is conferred by a wide variety of distinct sugar-specific permeases, the Enzymes II (EII), which are composed of one to four hydrophobic domains (domains A-D). Thus, these enzymes are responsible for the transport of carbohydrate across the bacterial membrane and for its phosphorylation [57].
The most important representative and better studied is the glucose-specific PTS that mediates the uptake and phosphorylation of this important sugar (Figure 5). The mechanism begins with the transfer of a phosphoryl from PEP to EI, the PEP-dependent protein-kinase, starting the phosphorylation cascade. This phosphate is subsequently transferred from EI-phosphate to HPr, which becomes phosphorylated. HPr-EI-phosphate, passes the phosphoryl group to the soluble EIIAGlc (encoded by the crr gene, part of the ptsHIcrr operon) and, in the final step, EIIAGlc-phosphate transfers the phosphoryl group to the glucose-specific membrane permease, EIICBGlc, encoded by the ptsG gene, which incorporates and phosphorylates glucose. These phosphotransfer reactions between PEP and EIIB domain of any sugar-specific EII are reversible. On the contrary, the final transfer of the high-energy phosphate group to the substrate is virtually irreversible [57, 59].
The PTS was initially associated with sugar transport and phosphorylation, controlling preferential use of carbon sources and the uptake systems of these energetically preferred sugars in bacteria, as explained above [57]. However, the PTS has been more recently recognized as a global regulator of the bacterial behaviors and metabolic processes.
Previous works revealed that many metabolic and regulatory functions are assigned to the components of the PTS. For instance, the regulation of flagellar motility or the movement towards the carbon sources – chemotatic response to PTS sugars – by unphosphorylated EI; the control of the net production of carbon and energy storage sources (glycogen [60] and
10 poly-β-hydroxybutyrate [61]); the transition between different metabolisms (fermentative and respiratory); the growth in a biofilm by EI [62]; the transport of non-carbon-compounds [63, 64] and also the regulation of the transport of non-PTS sugars [65] as well as the utilization of alternative carbon sources through the mechanisms of inducer exclusion and catabolite repression by EIIAGlc [57, 66].
Figure 5 – The glucose phosphoenolpyruvate-dependent phosphotransferase system. The
Phosphoenolpyruvate (PEP) starts the phosphorylation cascade by transferring its phosphoryl group to EI, which becomes phosphorylated (EI~P). EI~P passes its phosphate to HPr. HPr~P transfers the
phosphoryl group to EIIAGlc, which becomes phosphorylated (EIIAGlc~P). EIIAGlc~P phosphorylates
EIIBGlc. EIIBGlc~P passes the phosphate to the incoming glucose, as the sugar passes through the
pore created by the EIIC domain (adapted from reference [67]).
Additionally, the EIIAGlc was identified as the central processing unit of carbon metabolism in enteric bacteria, carrying out numerous regulatory functions. Specifically, unphosphorylated EIIAGlc represses adenylate cyclase and interacts with several non-PTS sugar permeases (lactose, maltose, melibiose), blocking the respective sugar incorporation and also binds to glycerol kinase to inhibit its activity (reviewed in [57]).
Given that PTS presents these regulatory roles and are capable of phosphorylating and interacting with numerous non-PTS proteins and thereby controlling their activity, it is not completely surprising that this system might play a crucial role in AI-2 incorporation and in Lsr transporter expression, as previously observed for mutants in the ptsI gene, that do not internalize AI-2 and do not activate the expression of the lsr operon [28]. The fact that AI-2 internalization is dependent on the AI-2-induced Lsr transporter, as well as on the Enzyme I
11 of the PTS, suggests that the bacterial integrates the information about its physiological state and according to that, regulates AI-2 signal incorporation.
Despite these new findings, regarding the involvement of PTS in AI-2 mediated regulation through the Lsr transport system, the molecular mechanism enabling the PTS-regulated transporter and thus the mechanisms linking the two systems are not understood.
In this work several questions related to PTS role in Lsr regulation were studied, specifically, the importance of the characteristic phosphotransferase activity of EI of the PTS, in the process of AI-2 incorporation and lsr operon activation was addressed. Another subject that was under the scope of this study was the role played by an important regulator of the PTS, the Mlc protein, in this mechanism of AI-2 internalization and in the expression of the Lsr transport system.
Most importantly, to determine the molecular mechanism involved in the regulation of AI-2 incorporation by EI, a genetic screen was also performed. The most important objective of this screen was the identification of suppressors of the ptsI mutation capable of internalizing AI-2. This screen has proven to be useful in identifying suppressors of the ptsI mutant phenotype because regulators of the system, previously identified, were scored as positive hits. The other hits still need to be analyzed to confirm their phenotypes and to select the most interesting mutants, for studying their role in the interaction between the PTS and the Lsr system.
Furthermore, in this study, a new tool to investigate the Lsr regulation was implemented: transcriptional fusions, of the lsr and lsrR promoters with the gene encoding for the green fluorescent protein (gfp), were constructed and their expression determined in several genetic backgrounds. These promoter-gfp fusions were compared with the commonly used promoter-lacZ fusions and their benefits and drawbacks were accessed.
In summary, the main objective of this research work was to gain a deeper knowledge of the AI-2/Lsr system, seeking to understand how the Lsr transporter is dependent on the PTS and reveal the molecular mechanism through which information about the physiological state of bacteria and regulation of AI-2 signal uptake is integrated.
12
2. Materials and Methods
2.1. Bacterial strains and growth conditions
The strains that were used in this research work are listed in Table 1. Wild-type (WT)
Escherichia coli K-12 strain MG1655 [68] was used as the parental strain for all the
subsequent genetic manipulations. The strains were grown with aeration at 37ºC, in Luria-Bertani (LB) broth supplemented with 100 mM MOPS buffer pH7 (LB MOPS), except where otherwise mentioned. When necessary, 1 μM of Isopropyl beta-D-1-thiogalactopyranoside (IPTG) or different concentrations of synthetically produced AI-2 [69] were supplied to the media at the time of inoculation. Where indicated below, medium was supplemented with antibiotics at the following final concentrations: Chloramphenicol, 25 mg.L-1 and Kanamycin, 50 mg.L-1.
2.2. β-galactosidase assays
Overnight cultures of Escherichia coli strains diluted 1:100 into fresh LB MOPS medium and grown with aeration at 37ºC to the OD600 indicated. Cells from 1 ml of culture were
harvested and resuspended in 1 ml of Z-Buffer 1X for determination of the β-galactosidase activity as described previously [70]. β-galactosidase activity was calculated as follows (OD420.min-1 x dilution factor)/OD600. All assays were performed in triplicate and were reported
as the mean β-galactosidase activity from triplicated data and error bars represent the standard error.
2.3. AI-2 activity assay
To measure AI-2 extracellular activity in Escherichia coli cultures, overnight cultures were diluted (1:100) into fresh LB MOPS medium in Erlenmeyer flasks and grown with aeration at 37ºC for the indicated time. Aliquots were collected at the times indicated and used to analyze the optical density at 600 nm (OD600) and to prepare cell-free culture fluids. The AI-2
detection and quantification in the cell-free culture fluids was measured using a LuxP-fluorescence resonance energy transfer (FRET) assay as previously described [71]. Cell-free culture fluids were prepared by filtration of liquid cultures through 96-well filtration plates. For the determination of AI-2 concentration, 2.5 μl of the cell-free fluid was added to 280 μl of purified CLPY FRET protein to a final concentration of 0.0125 mg/ml diluted in phosphate buffer. Results were compared to a calibration curve obtained from CLPY response to known concentrations of synthetic AI-2 prepared dilutions. Each sample was assessed in triplicates.
13
2.4. Plasmid construction
The plasmids pPBC01, pPBC03 and pPBC06 were used to study the expression of different promoters (lsrACDBFG promoter, lsrR promoter and the putative lsrK promoter) using a reporter green fluorescent protein (GFP). For cloning purposes, the DNA fragment containing the lsrACDBFG promoter, the DNA fragment containing the lsrR promoter and the DNA fragment containing the putative lsrK promoter were amplified by polymerase chain reaction (PCR) from the chromosome of Escherichia coli MG1655, respectively, using two pairs of primers (lsr1-GFP; lsr2-GFP; lsrR1-GFP; lsrR2-GFP; lsrK1-GFP; lsrK2-GFP) and the Bio-X-Act DNA polymerase (Bioline), to ensure great accuracy, during the amplification. The products of PCR were then digested with SalI and SphI, and subsequently were ligated with
SalI and SphI-digested pCMW1 where the gfp transcriptional fusions were constructed. Each
transcriptional fusion was cloned into the plasmid pCMW1, upstream of the promoterless gfp gene. The constructed plasmids were transformed into Escherichia coli MG1655. All plasmid constructs were confirmed by DNA sequencing carried out by the IGC sequence facility. The plasmids and the sequences of all primers used in this study are listed in Table 2 and Table 3, respectively.
2.5. DNA manipulations
All DNA manipulation was performed using standard procedures. T4 DNA ligase and restriction enzymes were obtained from New England Biolabs (NEB). The transformation of
Escherichia coli with the plasmids used in this study was performed in 0.2-cm electroporation
cuvettes, using a Bio-Rad Micro Pulser. The plasmids EI and the plasmid pQELL-EIH189A were introduced into the strain CSP108 and the pQE32-lacIq plasmid was introduced into the strains CSP108 and AS8. Each of the plasmids pPBC01, pPBC03 and pPBC06 was introduced into the following strains by electroporation: MG1655, KX1448, KX1228 and AS13. The vector used for construction of all the promoter-gfp fusions was the plasmid pCMW1. In the absence of insert DNA, the pCMW1 plasmid shows no detectable gfp expression. All strains and plasmids used in this study are listed in Table 1 and Table 2, respectively.
2.6. Green fluorescent protein expression assay
Cultures of the strains were grown overnight in Brain Heart Infusion (BHI) medium, diluted 1:100 into fresh BHI medium supplemented with chloramphenicol (25 mg.L-1) and grown at 37º C with aeration. At different time points after inoculation, culture samples of 500 μl were collected for the determination of the OD600 and green fluorescent protein (GFP) expression.
14 The cells were harvested and resuspended in 500 μl of Phosphate Buffered Saline (PBS) buffer 1X, to mitigate the fluorescence from the growth medium. GFP production of each sample was measured in a black clear bottom plate using the Victor3 multilabel counter (Wallac). Each sample was assessed in triplicates and the strain containing the promoterless GFP plasmid pCMW1 was also measured to exclude the background fluorescence. Fluorescence units produced by a strain were defined as [GFP fluorescence (strain)/OD (strain)] – [GFP fluorescence (pCMW1)/OD (pCMW1)].
2.7. Construction of the strains carrying the lsrR-lacZ promoter fusion
To construct the strains carrying the lsrR-lacZ promoter fusion, we used the P1 vir lysate from the strain KX1267 to transduce the lsrR-lacZ fusion, linked to a kanamycin resistance cassette, into four strains with distinct genetic backgrounds MG1655 (WT), KX1448 (ΔlsrK), KX1228 (ΔluxS) and AS13 (ΔlsrR) and selected for growth on LB agar supplemented with kanamycin, 50 mg. L-1. Next, a ΔcyaA::Cm deletion was transduced to each the previous strains. For that, we used a P1 vir lysate from AS2 (ΔcyaA::Cm) and the new transductants were selected in LB agar supplement with kanamycin, 50 mg.L-1 and chloramphenicol, 25 mg.L-1. For the constructed strains to became insensitive to catabolite repression we transduced a P1 bacteriophage lysate from the strain RD14 which contains a cyaA deletion linked to kanamycin and the crp* encoding a derivative of catabolite activator protein (CAP) that acts as a transcriptional activator in the absence of cyclic AMP (cAMP). Because a ΔcyaA::Cm mutant cannot grow using glycerol, the crp* gain of function mutation was selected by growth on M63 agar medium containing supplemented with glycerol.
2.8. Screen for regulators of EI of the PTS and suppressors of the ptsI mutation
To identify suppressors of the ptsI mutant strain, capable of internalizing AI-2 and activating lsr operon expression, as well as regulators of EI of the PTS, a genetic screen was performed. All the single-gene deletions of all nonessential genes in Escherichia coli present in the Keio collection library (3985) were transduced to the strain CSP108 (lsr-lacZ, ΔcyaA,
crp*, ΔptsI) carrying a ptsI deletion and a lsr-lacZ reporter fusion. To identify target genes, we
screened the new transductants strains that showed lsr-lacZ expression higher than the parent strain.
15
2.8.1. Preparation of P1 bacteriophage lysates in 96-well plates
Overnight cultures of the all the strains of the Keio collection were grown in 96-well plates in LB at 30ºC with aeration. These overnight cultures were diluted into LB supplemented with 0,2% glucose and 5 mM CaCl2 and the 96-well plate was incubated at 37ºC during 1 hour
shaking. To each well, P1 bacteriophage lysate of the wild-type strain, MG1655, was added and the plate was incubated for 4 hours, at 37ºC with shaking, until the cells lyse. The plate was centrifuged and the supernatant was transferred to a new 96-well plate. In the final step, chloroform was added to each well, to eliminate the remaining cells and for storage of the lysates. The whole procedure was performed for all the 3985 single deletions of the Keio collection library [72].
2.8.2. P1 transduction in 96-well plates
As mentioned above, all the single deletions of the Keio collection were transduce into the
ptsI deletion strain, CSP108 (lsr-lacZ, ΔcyaA, crp*, ΔptsI). First, overnight cultures of the
recipient strain were grown at 30ºC in LB supplemented with 10 mM MgSO4 and 5 mM
CaCl2. The 96-well plates were filled with overnight culture, the respective P1 bacteriophage
lysates were added to each well and the plates were incubated for 45 min at 37ºC without shaking. LB supplemented with 50 mM Na Citrate was added to each well and the plates were incubated for 2h30min, at 37ºC without shaking. Next, LB supplemented with kanamycin, 100 mg.L-1, was supplied to each well and the plates were incubated overnight at 37ºC, with aeration. Next day, using a replica plater, a stamp of each 96-well plate, containing the transductants, was done to LB agar plates supplemented with kanamycin 30 mg. liter-1 plus 30 mM Na Citrate.
2.8.3. Screening using MacConkey Lactose Agar plates
The obtained transductant strains were plated in MacConkey lactose agar plates, using a replica plater. The plates were incubated at 37ºC and the phenotype of each transduction was assessed. The results of the genetic screen were further processed and analysed.
16
Table 1 – Escherichia coli strains used in this study.
Strain Relevant genotype Source
AS4 lsr-lacZ, ΔcyaA, crp*, ΔluxS Lab collection AS8 lsr-lacZ, ΔcyaA, crp* Lab collection AS2 ΔcyaA::Cm Lab collection
AS13 ΔlsrR Lab collection
AS82 lsr-lacZ, ΔcyaA, crp*, ΔlsrK Lab collection
CSP108 lsr-lacZ, ΔcyaA, crp*, ΔptsI Lab collection
CSP370 lsr-lacZ, ΔcyaA, crp*, Δmlc::Kan Lab collection
KX1228 ΔluxS Lab collection
KX1267 lsrR-lacZ Lab collection
KX1448 ΔlsrK Lab collection
MG1655 Wild-type strain Lab collection
PBC02 MG1655 carrying the plasmid pPBC01 This study
PBC03 MG1655 carrying the plasmid pPBC03 This study
PBC06 MG1655 carrying the plasmid pPBC06 This study
PBC07 Strain CSP108 carrying the plasmid pQE32-lacIq This study
PBC09 Strain CSP108 carrying the plasmid pQELL-EI This study
PBC19 Strain AS8 carrying the plasmid pQE32-lacIq This study
PBC21 Strain KX1448 carrying the plasmid pPBC01 This study
PBC23 Strain KX1448 carrying the plasmid pPBC03 This study
PBC25 Strain KX1448 carrying the plasmid pPBC06 This study
PBC27 Strain KX1228 carrying the plasmid pPBC01 This study
PBC30 Strain KX1228 carrying the plasmid pPBC03 This study
PBC31 Strain KX1228 carrying the plasmid pPBC06 This study
PBC35 Strain CSP108 carrying the plasmid pQELL-EIH189A This study
PBC41 Strain AS13 carrying the plasmid pPBC01 This study
PBC44 Strain AS13 carrying the plasmid pPBC03 This study
PBC45 Strain AS13 carrying the plasmid pPBC06 This study
PBC105 lsrR-lacZ, cyaA::Cm, crp* This study
PBC107 lsrR-lacZ, cyaA::Cm, crp*, ΔluxS This study
PBC109 lsrR-lacZ, cyaA::Cm, crp*, ΔlsrK This study
17
PBC148 lsr-lacZ, ΔcyaA, crp*, ΔptsI, Δmlc::Kan Lab collection
Table 2 – Plasmids used in this study.
Plasmid Relevant genotype or property Source
pPBC01 pCMW1 derivative, lsr promoter-GFP transcriptional fusion, CmR
and KanR
This study
pPBC03 pCMW1 derivative, lsrR promoter-GFP transcriptional fusion, CmR
and KanR
This study
pPBC06 pCMW1 derivative, lsrK promoter-GFP transcriptional fusion, CmR
and KanR
This study
pQE32-lacIq Vector [73]
pQELL-EI EI expression vector with IPTG inducible promoter [73]
pQELL-EIH189A EIH189A expression vector with IPTG inducible promoter [73]
Table 3 – Primers used in this study.
Primer name Oligonucleotide sequence (5’-3’)
lsr1-GFP ACGGCATGCGAGTTTCATATTCCAGACAGCCTTC lsr2-GFP ACGGTCGACGAACTGGCGTTAATCTGACGTAG lsrK1-GFP ACGGCATGCGAACTGGCGTTAATCTGACGTAG lsrK2-GFP ACGGTCGACGAGTTTCATATTCCAGACAGCCTTC lsrR1-GFP ACGGCATGCATGCTGGCGATTGGTTTTGGCGAG lsrR2-GFP ACGGTCGACATGCCGCCACTCCGCCTGTCCCACT
18
3. Results
3.1. The role of EI of PTS in AI-2 internalization
The phosphoenolpyruvate-dependent phosphotransferase system (PTS) is a mechanism responsible for the incorporation of a wide variety of carbohydrates through the bacterial cell membrane. This process occurs with concomitant phosphorylation of the imported sugar and uses phosphoenolpyruvate (PEP) as the donor of the phosphoryl group that will trigger the phosphorylation cascade [57]. This is composed of several proteins that mediate the transfer of the phosphate from PEP to the internalized sugar (Figure 5).
Although, the PTS is mainly associated with the incorporation and the phosphorylation of different carbohydrate sources and with the control of the preferential use of these sugars and their respective uptake systems [57]; several metabolic and regulatory functions were attributed to the proteins that compose the PTS.
Previous results showed that the PTS plays an in important function in the AI-2 mediated regulation in Escherichia coli. Specifically, it was shown that a deletion mutant in the gene
ptsI, that encodes for the EI of PTS, do not incorporate AI-2 neither activate the expression
of the lsr operon [28].
3.1.1. A functional EI is required for AI-2 incorporation and lsr operon expression
The capacity of the PTS to incorporate and concomitantly phosphorylate the internalized sugars relies on the phosphotransferase activity of the several proteins that compose the system. Specifically, the phosphotransferase activity of EI is crucial to remove the phosphoryl group from PEP and to initiate the phosphorylation cascade that characterizes the PTS. Furthermore, this phosphotransferase activity of EI is highly indispensable to accomplish the several functions that are attributed to this enzyme but there are exceptions. Specifically, phosphorylation of EI is not necessary for the activation of the regulator of the β-glucoside utilization system in Escherichia coli [73].
As shown in previous studies [28], the PTS-dependent removal of AI-2 from the extracellular medium still requires the LsrK kinase because in a lsrK mutant extracellular levels of AI-2 are high and no internalization is observed. So, in this work it was determined whether the characteristic phosphotransferase activity of the EI is crucial for AI-2 incorporation and for activation of the lsr operon expression.
To determine AI-2 internalization in different mutants, the extracellular AI-2 activity was determined using the LuxP-fluorescence resonance energy transfer (FRET) assay [71] which allows the quantification of AI-2 concentration in the culture fluids of the studied strains.
19 Furthermore, the respective expression of the lsr operon was determined by measuring the transcription of a lsr-lacZ promoter fusion inserted at the lambda attachment site.
As already established and also shown in Figure 6A, the extracellular AI-2 activity in a wild-type strain peaks as bacteria divide in exponential growth phase and rapidly declines due to the expression of the Lsr transport system that incorporates AI-2. After 4 hours of growth, corresponding to a certain AI-2 concentration in the medium and to a certain number of bacterial cells; AI-2 is almost completely depleted from the extracellular medium. In contrast, in the deletion mutant of the ptsI gene, encoding for EI of PTS, there is a normal production of AI-2 and its concentration rises as bacteria grow but AI-2 is not incorporated and the extracellular AI-2 activity continues to increase. Induction of the lsr operon reflects the levels of AI-2 internalization observed for the transcription of the lsr-lacZ promoter fusion (Figure 6B): the wild-type strain shows activation of lsr operon expression since AI-2 is being internalized and thus induces transcription of the Lsr transporter, while the mutant in the ptsI gene does not activate the expression of the lsr operon. These data show that EI is important for Lsr regulation as stated in previous studies [28].
Figure 6 – EI from PTS and its phosphotransferase activity are required for AI-2 internalization and lsr operon transcription. The extracelular AI-2 activity A) and transcription of the lsr operon at
OD600=4 B) were measured in the following strains: WT + vector [PBC19: lsr-lacZ, ΔcyaA, crp* +
pQE32-lacIq]; ptsI + vector [PBC07: lsr-lacZ, ΔcyaA, crp*, ΔptsI + pQE32-lacIq]; ptsI + p(EI-WT)
[PBC09: lsr-lacZ, ΔcyaA, crp*, ΔptsI + pQELL-EI] and ptsI + p(EI-H189A) [PBC35: lsr-lacZ, ΔcyaA,
crp*, ΔptsI + pQELL-EIH189A]. The lsr-lacZ transcription was measured at OD600=4 because it
corresponds to the maximum lsr activation in the wt strain. Error bars represent standard deviation of triplicates.
20 To address if the phosphotransferase activity of EI is required for AI-2 incorporation and
lsr operon expression, the mutant ptsI strain was tested for complementation with a wild-type ptsI gene or with a ptsI mutant allele that encodes for an EI mutated in the phosphorylation
site (EI-H189A). Each gene was introduced into the ptsI mutant strain in a multicopy plasmid and transcribed under control of an IPTG-inducible promoter. The extracellular AI-2 activity and the expression of the lsr-lacZ promoter fusion were accessed for each strain.
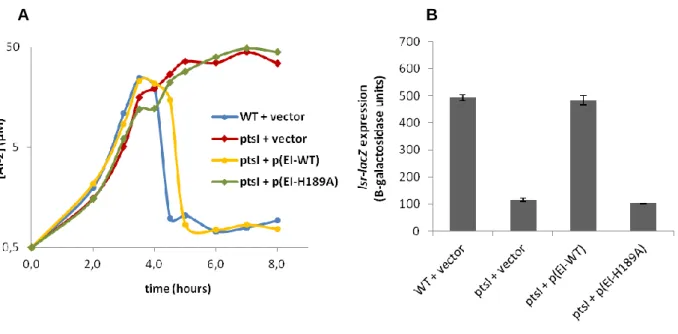
As shown in Figure 6, when the wild-type allele of ptsI is provided to the ptsI mutant the wild-type phenotype is recovered and the impairment in AI-2 internalization and lsr operon transcription of the ptsI mutant is overcome (ptsI + p(EI-WT)). A slight delay in AI-2 incorporation is still observed but this is due to the intrinsic growth defect that the ptsI mutant presents even when it is complemented with the wild-type ptsI allele (Figure 7).
On the other hand, when the ptsI mutant expresses the gene encoding EI, with the mutated phosphorylation site, EI-H189A (the histidine residue has been replaced by an alanine); no incorporation of AI-2 and no properly activation of the transcription of the lsr operon were observed. Together, these data show that a functional EI, with preserved phosphotransferase ability is required for AI-2 incorporation and to activate lsr operon transcription.
Figure 7 – Growth curves of the different studied strain. OD600 was measured in the following
strains: WT + vector [PBC19: lsr-lacZ, ΔcyaA, crp* + pQE32-lacIq]; ptsI + vector [PBC07: lsr-lacZ,
ΔcyaA, crp*, ΔptsI + pQE32-lacIq]; ptsI + p(EI-WT) [PBC09: lsr-lacZ, ΔcyaA, crp*, ΔptsI + pQELL-EI]
and ptsI + p(EI-H189A) [PBC35: lsr-lacZ, ΔcyaA, crp*, ΔptsI + pQELL-EIH189A
21
3.1.2. Mlc, an important regulator of PTS, does not play a role in Lsr regulation
Mlc is a major regulator which plays an important function in PTS-dependent transport of glucose. This regulator is responsible for repressing the expression of the ptsG gene that encodes for the glucose-specific permease EIIBCGlc and also the gene ptsI and ptsH, that encode for the general components of the PTS, designated EI and HPr, respectively.
In the absence of glucose, Mlc interacts with the unphosphorylated form of EIIBCGlc, which sequesters that regulator preventing its mediated repression and as a consequence transcription of the PTS components increases.
Mlc is also associated with the repression of the manXYZ genes that encode for the PTS proteins for the transport of mannose and with the repression of the transcriptional activator MalT for the mal regulon. By repressing MalT, Mlc indirectly represses the malEFG, malK,
lamB and malPQ operon, which encode genes responsible for the transport and catabolism
of maltose, a non-PTS sugar [57, 67, 74]. According to this, Mlc is responsible for repressing the transcription of permeases that perform the transport of the PTS sugars, glucose and mannose, as well as the expression of the transporter of non-PTS sugars, such as maltose. Thus, here we tested if Mlc could play a direct function in AI-2 incorporation by regulating one of the AI-2 permeases directly or indirectly via EI.
In order to address this hypothesis, the extracellular AI-2 activity and the expression of the
lsr-lacZ transcriptional fusion were determined in a mlc null background and also in a double
mutant in the genes ptsI and mlc (Figure 8).
Figure 8 – Mlc does not play a role in AI-2 incorporation and lsr operon transcription. The
extracelular AI-2 activity A) and transcription of the lsr operon at OD600=4 B) were measured in the
following strains: wt [AS8: lsr-lacZ, ΔcyaA, crp*]; lsrK [AS82: lsr-lacZ, ΔcyaA, crp*, ΔlsrK]; ptsI
[CSP108: lsr-lacZ, ΔcyaA, crp*, ΔptsI); mlc [CSP370: lsr-lacZ, ΔcyaA, crp*, Δmlc::Kan) and ptsI, mlc
[PBC148: lsr-lacZ, ΔcyaA, crp*, ΔptsI, Δmlc::Kan]. The lsr-lacZ transcription was measured at OD600=4
because it corresponds to the maximum lsr activation in the wt strain. Error bars represent standard deviation of triplicates.





![Figure 7 – Growth curves of the different studied strain. OD 600 was measured in the following strains: WT + vector [PBC19: lsr-lacZ, ΔcyaA, crp* + pQE32-lacI q ]; ptsI + vector [PBC07: lsr-lacZ, ΔcyaA, crp*, ΔptsI + pQE32-](https://thumb-eu.123doks.com/thumbv2/123dok_br/15476036.1036054/30.892.248.627.637.940/figure-growth-different-studied-measured-following-strains-δcyaa.webp)