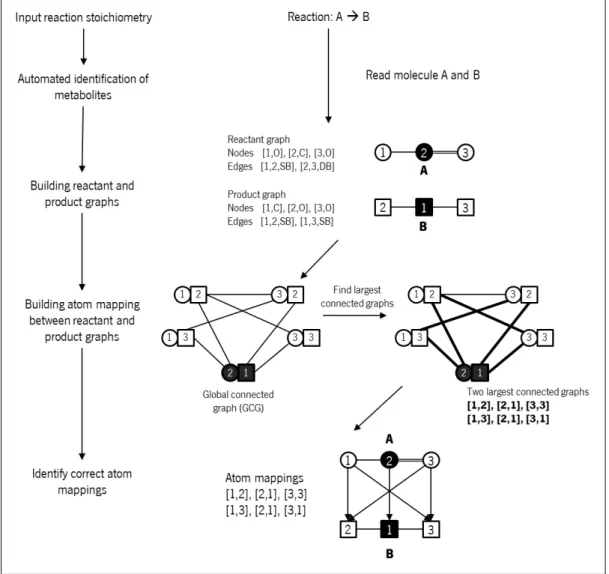
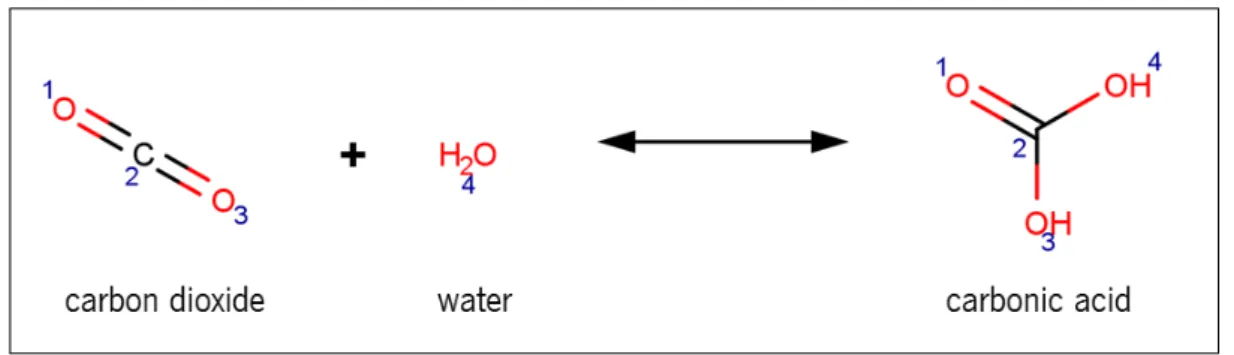
A critical evaluation of automatic atom mapping algorithms and tools
94
0
0
Texto
Imagem
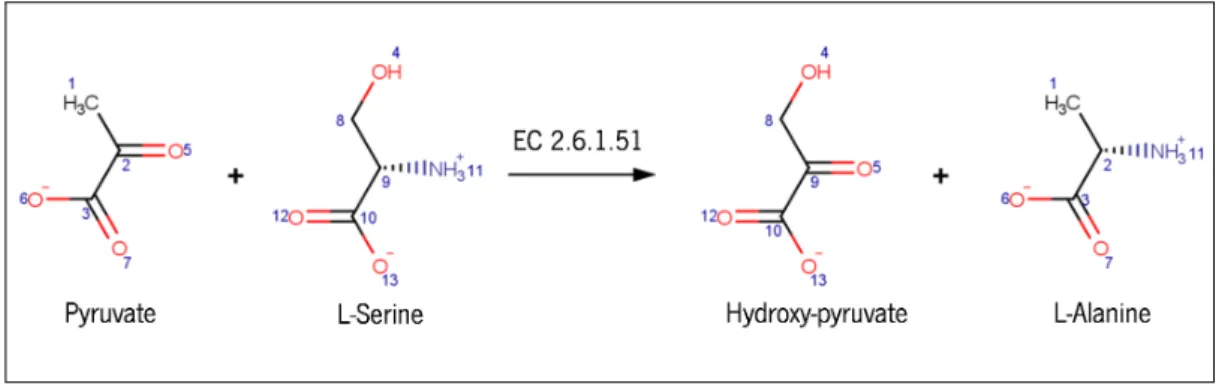
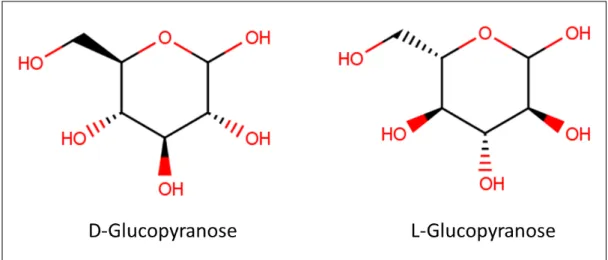
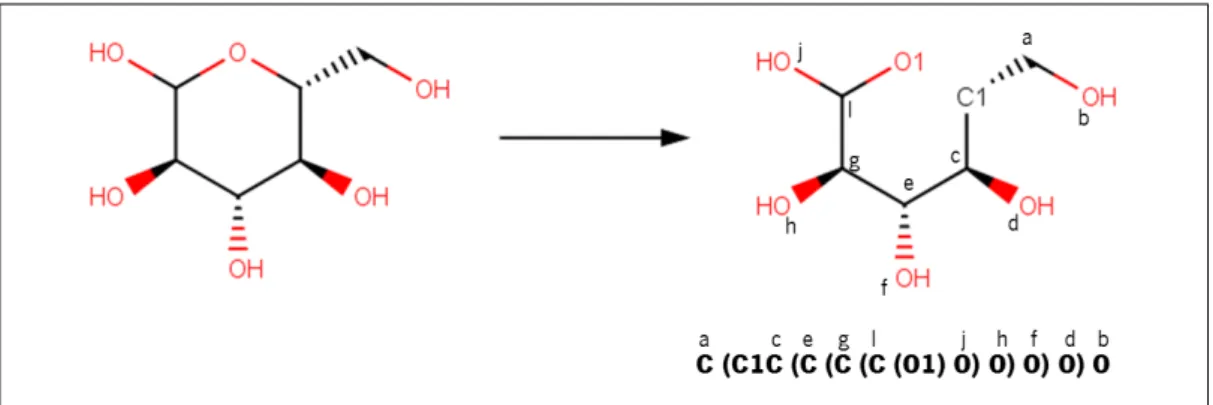
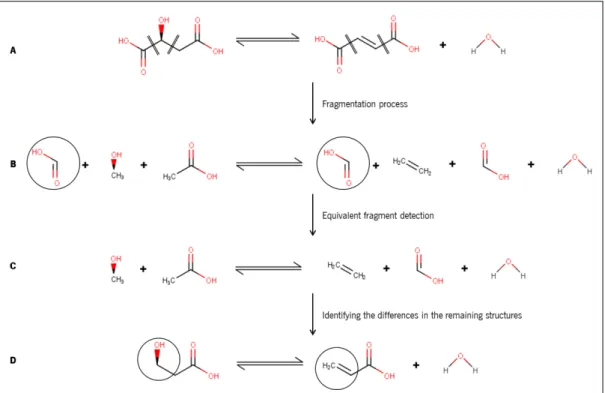
+7
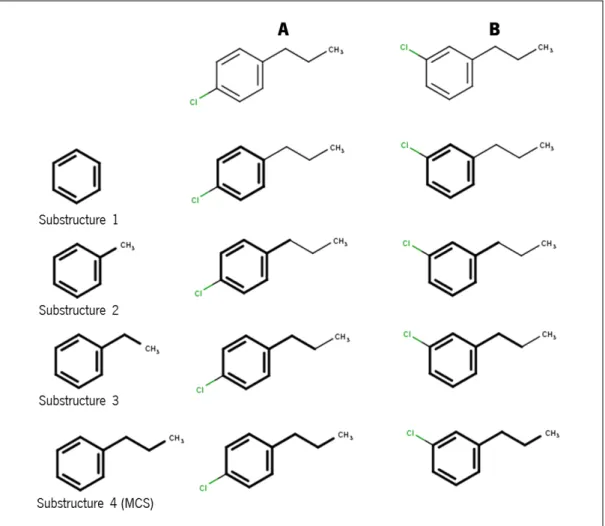
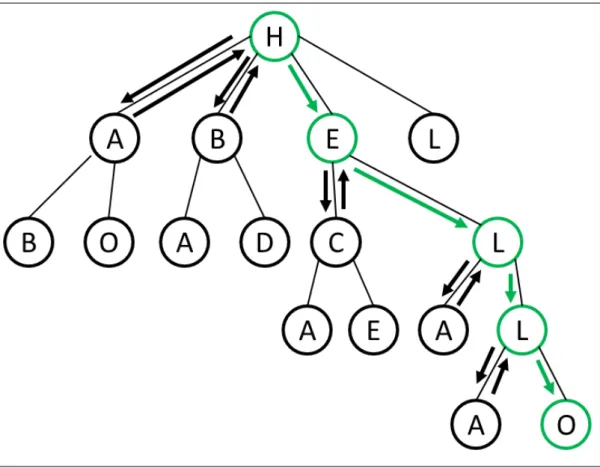
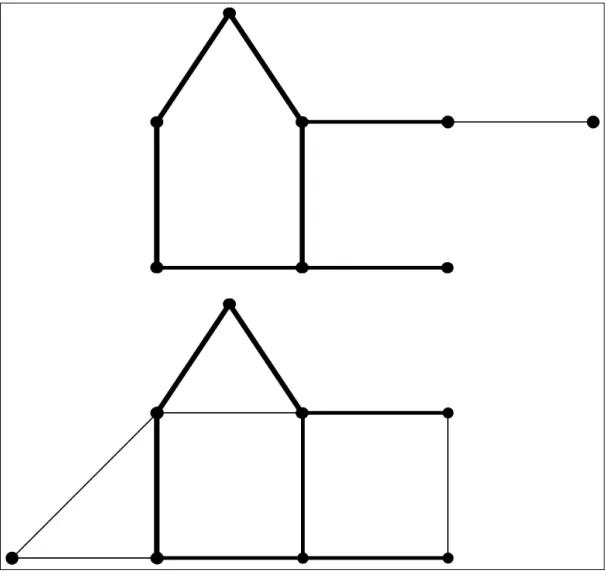
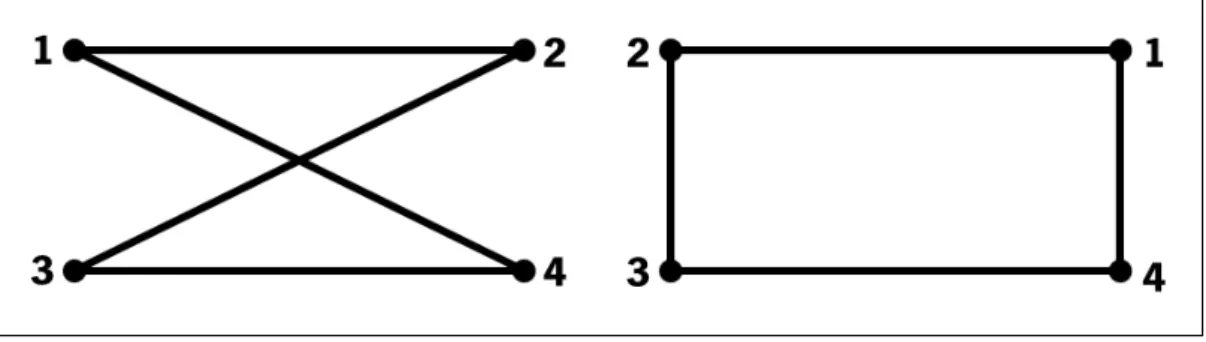
Documentos relacionados