REDEMAT
R
EDET
EMÁTICA EME
NGENHARIA DEM
ATERIAISUFOP – CETEC – UEMG
Dissertação de Mestrado
"Simulação computacional de misturas binárias
de nanotubos de carbono e anfifílicos em solução
aquosa via Método de Monte Carlo"
Autor: Leandro Lopes Hermsdorff
Orientador: Prof. Américo Tristão Bernardes
Co-orientador: Prof. Carlos Felipe Saraiva
Pinheiro
REDEMAT
R
EDET
EMÁTICA EME
NGENHARIA DEM
ATERIAISUFOP – CETEC – UEMG
Leandro Lopes Hermsdorff
"Simulação computacional de misturas binárias de nanotubos de
carbono e anfifílicos em solução aquosa via Método de Monte
Carlo"
Dissertação de Mestrado apresentada ao Programa de Pós-Graduação em Engenharia de Materiais da REDEMAT, como parte integrante dos requisitos para a obtenção do título de Mestre em Engenharia de Materiais.
Área de concentração: Análise e Seleção de Materiais
Orientador: Prof. Américo Tristão Bernardes
Catalogação: [email protected] H556s Hermsdorff, Leandro Lopes.
Simulação computacional de misturas binárias de nanotubos de carbono e anfifílicos em solução aquosa via Método de Monte Carlo [manuscrito] / Leandro Lopes Hermsdorff – 2009.
vii, 56f.: il. color.; grafs.; tabs.
Orientador: Prof. Dr. Américo Tristão Bernardes. Coorientador: Prof. Dr. Carlos Felipe Saraiva Pinheiro.
Dissertação (Mestrado) - Universidade Federal de Ouro Preto. Escola de Minas. Rede Temática em Engenharia de Materiais.
Área de concentração: Análise e Seleção de Materiais.
1. Nanotubos de carbono - Teses. 2. Simulação computacional - Teses. 3. Método de Monte Carlo - Teses. I. Universidade Federal de Ouro Preto. II. Título.
Agradecimentos
A
Agradeço ao Senhor Deus, cuja misericórdia se estende pelos séculos dos séculos. A Ele seja a honra, a glória e o louvor por cada conquista.
Agradeço aos meus pais, Vecão e Heloísa, pelo carinho e amor durante toda minha vida. Eles me são inspiração para permanecer na luta da vida. Todo trabalho duro e todo esforço empreendido permitiu que eu pudesse chegar até aqui. Não há um só dia em que a saudade não me visite para relembrar-me o quão preciosos são para mim.
Agradeço a meus irmãos Wevergton e Leilane pelo incentivo, confiança, amizade, “puxões de orelha” e por tornarem meus dias sobre a terra ainda mais valiosos.
Agradeço a Leciana pelo carinho, apoio e companhia nesses últimos dois anos. Tão pouco tempo com muita história pra contar. Preciosos e inesquecíveis dias...
Agradeço a todos irmãos da igreja Peniel Ouro Preto pelo carinho e amor por todos estes anos.
Agradeço ao Daniel e a Rose por aceitarem minha amizade e me fazer sentir “da família”. Nota 1000!!!
+Agradeço ao Hugo e ao Claudiomiro pelos momentos deCS. Figuraças!!!
-Planting the bomb!!!
Agradeço ao professor Américo Tristão Bernardes por me orientar e aturar todas inúmeras
ligações, emails e aturar meus momentos de desespero... Esse homem tem paciência! It’s a
super hero! The Capitain Américo!!!
Agradeço ao professor Dr. Romuel Machado por fazer minha vida cristã mais útil... Jesus te ama!
Agradeço aos demais professores por todas as dúvidas tiradas e por me aturarem em suas salas com perguntas e mais perguntas.
Ao Tiago e à Michele por todos “cafezinhos com piadas” na cantina do ICEB. Ao lembrar desses saudosos amigos me pesa muito o coração, visto que não estão mais entre nós... estão em São Paulo cuidando do doutorado e me deixaram para trás!!! Ôh falta de consideração!!!
Agradeço de novo a Michele por me aturar, aconselhar e mesmo cuidar no tempo que pas-samos juntos. Saudades.
Agradeço aos companheiros do laboratório de simulação do departamento de física da UFOP que têm me acompanhado, ajudado e aturado nesse período de minha vida. Grato pela consideração e pela preocupação; fatores esses que tornaram esses dias mais leves.
Agradeço encarecidamente a todos demais amigos “ufopinianos” e não “ufopinianos” ao qual devo favores e agradecimentos, cujos nomes provavemente não caberiam nas próximas 50 páginas mesmo usando letra tamanho 8... Valeu galera!
Por último, agradeço novamente à Deus dizendo:
“Compassivo e misericordioso é o Senhor... Não nos trata segundo os nossos pecados, nem
nos retribui segundo as nossas iniquidades... Pois é grande a sua benignidade para com os
que o temem.”
Salmo 103:8,10,11
Sumário
Lista de Figuras iii
Lista de Tabelas v
Resumo vi
Abstract vii
1 Introdução 1
2 Objetivos 3
3 Revisão Bibliogáfica 4
3.1 O Carbono e Seus Alótropos . . . 4
3.2 Nanotubos de Carbono . . . 6
3.2.1 Morfologia . . . 7
3.2.2 Nanotubos de carbono de paredes múltiplas . . . 11
3.3 Surfactantes . . . 12
3.3.1 Dodecil Sulfato de Sódio . . . 14
3.4 Comportamento em Solução Aquosa . . . 14
4 Metodologia 17 4.1 O Método de Monte Carlo . . . 17
4.1.1 Processos Estocásticos . . . 19
4.1.2 Cadeias de Markov . . . 20
4.1.3 O Algoritmo de Metrópolis . . . 21
4.2 Função de estrutura . . . 22
5 Modelo 25 5.1 Moléculas e Movimentos . . . 26
5.1.1 Molécula de Água (w) . . . 26
5.1.2 Molécula Anfifílica (surf) . . . 27
SUMÁRIO
5.1.2.2 Translação . . . 28
5.1.2.3 Rotação . . . 28
5.1.2.4 Pivô . . . 29
5.1.3 Nanotubo de Carbono (nc) . . . 30
5.1.3.1 Translação . . . 31
5.1.3.2 Rotação . . . 31
5.2 Hamiltoniana . . . 31
6 Resultados e discussões 34 6.1 Simulações das moléculas de água . . . 34
6.2 Simulações das moléculas anfifílicas na presença de água . . . 37
6.3 Simulações dos nanotubos de carbono na presença de moléculas anfifílicas em solução aquosa . . . 42
6.3.1 Resultado das simulações com 1 nanotubo . . . 44
6.3.2 Resultado das simulações com 2 nanotubos . . . 48
6.3.3 Resultado das simulações com 3 nanotubos . . . 49
7 Conclusões 51
Lista de Figuras
3.1 Estrutura do grafite. . . 4
3.2 Célula unitária do diamante. . . 5
3.3 FulerenoC60. Figura extraída de http://lqes.iqm.unicamp.br, em 29/04/08. . . . 6
3.4 Representação de uma folha de grafite . . . 8
3.5 Tipos de nanotubos segundo a quiralidade. . . 9
3.6 Comprimento da célula unitária segundo a quiralidade. . . 10
3.7 Nanotubos de parede simples e de paredes múltiplas . . . 11
3.8 Representação de uma micela. . . 13
3.9 Representação esquemática de uma lamela. . . 13
3.10 Variação da tensão superficial da água em função da concentração de moléculas detergentes. . . 14
3.11 Esquemas representativos da molécula do SDS . . . 14
4.1 Algoritmo de Metropolis. . . 23
5.1 Modelo de partículas em rede e livre de rede. . . 25
5.2 Esquema ilustrativo do movimento de reptação . . . 28
5.3 Esquema ilustrativo do movimento de translação. . . 28
5.4 Esquema ilustrativo do movimento de rotação. . . 29
5.5 Esquema ilustrativo do movimento de pivô. . . 29
5.6 Número de pontos em um mesmo plano para três tipos de nanotubos. . . 30
5.7 Potenciais entre as partículas. . . 33
6.1 Energia em função do tempo de MC de um sistema com 400 águas em uma caixa de tamanho 40 e temperatura T =0,5. O destaque mostra a região onde ocorreu o encontro das curvas de energia das simulações . . . 35
6.2 400 águas em uma caixa quadrada 20×20 com T =0,5. À esquerda: con-figuração inicial de maior energia. No centro: concon-figuração inicial de menor energia. À direita: amostragem da configuração de equilíbrio (configuração “final”). . . 35
LISTA DE FIGURAS
6.5 Energia em função do tempo de MC em temperaturas T =1 (em cima, à
es-querda) e T =2 (em cima, à direita). A figura de baixo corresponde a uma
amostragem do sistema no equilíbrio em temperaturaT =2. . . 37
6.6 . . . 38 6.7 Configuração inicial (acima) e final (abaixo) para um sistema contendo 40
an-fifílicos e 2380 águas na tempertaturaT =3. Os círculos brancos representam
as águas. Os círculos pretos e cinzas representam, respectivamente, as cabeças e as caudas dos anfifílicos. . . 39 6.8 Detalhe da Figura6.7. . . 40 6.9 CurvaX1f reeversusXamph para diferentes temperaturas. . . 41
6.10 Configuração típica do equilíbrio térmico. Dados: 80 anfifílicos, 2260 águas, T =3,0. . . 42
6.11 Detalhe da Figura6.10. . . 42 6.12 Configuração típica do equilíbrio térmico. Dados: 140 anfifílicos, 2080 águas,
T =3,0. . . 43
6.13 Detalhe da Figura6.12. . . 43 6.14 Energia em função do números de PMC. Dados: 1 nanotubo, 200 anfifílicos,
1890 águas,T =3,0. . . 44
6.15 Função distribuição radial de uma configuração de equilíbrio para 20, 40, 60 e 80 anfifílicos em solução com um nanotubo. As figuras estão dispostas em sentido da escrita . . . 45 6.16 Função distribuição radial do sistema no equilíbrio para 20, 40, 60 e 80
anfifí-licos em solução com um nanotubo obtidas a partir da média de 40 amostras. Novamente, as figuras estão dispostas em sentido da escrita . . . 46 6.17 Retrato da configuração de equilíbrio das simulações com diferentes
concentra-ções de equilíbrio: 20 anfifílicos (superior esquerda), 120 anfifílicos (superior direita), 160 anfifílicos (inferior esquerda) e 200 anfifílicos (inferior direita).
Imagem obtida após 1,5M PMC. Novamente, as figuras estão dispostas em
sentido da escrita . . . 47 6.18 Retrato da configuração de equilíbrio das simulações de sistemas com dois
na-notubos com diferentes concentrações de equilíbrio: 60 anfifílicos (superior esquerda), 100 anfifílicos (superior direita) e 180 anfifílicos (abaixo). Imagem obtida após 1,5MPMC. As figuras estão dispostas em sentido da escrita . . . . 49 6.19 Retrato da configuração de equilíbrio das simulações de sistemas com três
Lista de Tabelas
5.1 Possíveis valores para E0. A intensidade da repulsão entre duas moléculas
quaisquer com ri j ≤d é∞(distância de exclusão). A intensidade da repulsão
Resumo
Abstract
Capítulo 1
Introdução
À medida que o conhecimento em nanociências se desenvolve, suas múltiplas possibilidades de aplicação estimulam a imaginação e abrem perspectivas promissoras de inovação tecnológica. Ao longo das últimas décadas, parte da comunidade científica tem estado envolvida no estudo do mundo nanométrico, investigando as propriedades únicas e inesperadas dos nanomateriais, assim como suas potenciais aplicações em ciência e tecnologia [1]. Nesse sentido, muitos esforços têm sido realizado com o intuito de atingir o tão desejado controle em nível atômico e molecular sobre os processos industriais.
As nanociências reúnem frequentemente diferentes domínios possuindo uma abordagem multidisciplinar e exigindo enormes esforços de investigação fundamental e aplicada. De fato, esses domínios envolvem uma diversidade de especialidades esperando-se que resultem em inovações que possam contribuir para a resolução de diversos problemas que a sociedade tem enfrentado, tais como as aplicações médicas, as tecnologias da informação, a produção e arma-zenamento de energia, a instrumentação para o estudo de propriedades da matéria, as ciências da vida, as tecnologias da comunicação, os transportes, o ambiente e também a área aeroespacial.
Dentre os materiais de interesse atualmente encontram-se os nanotubos de carbono. Cer-tamente esse material tem atraído maior interesse pelas suas surpreendentes propriedades me-cânicas, térmicas e eletrônicas, entre outras [2]. A compreensão dessas propriedades, assim como o desenvolvimento de processos de fabricação viáveis do ponto de vista econômico, são de fundamental importância para que os nanotubos de carbono possam ser utilizados em suas mais diversas aplicações. Desde 2001, em sua descoberta [3], até o fim de 2006 estima-se que foram publicados aproximadamente 19000 trabalhos envolvendo esse material [4]. Entretanto, sua aplicação é ainda limitada devido ao problema de insolubilidade em praticamente todos os solventes [1]. A agregação dos nanotubos provoca a redução das propriedades mecânicas e elé-tricas dos tubos individuais, tornando um obstáculo à suas aplicações tecnológicas. Isso, aliado a dificuldade de manipular nanotubos em feixes, têm sido motivação para o desenvolvimento de novos métodos visando promover a solubilização, dispersão e separação dos tubos [5].
di-ferentes comprimentos, quiralidade e diâmetros [6]. Dessa forma, observa-se que o desenvol-vimento de sistemas de engenharia macroscópicos está ainda em estágio inicial, ou seja, para viabilizar possíveis aplicações comerciais é ainda necessário a utilização de uma rota de pro-cessamento única e inovadora [7]. Os nanotubos devem ser modificados para se conseguir solubilidade e integração em estruturas ordenadas e diversos métodos tem sido propostos para atingir esse objetivo.
Esse trabalho é organizado da seguinte forma. No Capítulo 3 é apresentado uma breve
Capítulo 2
Objetivos
Capítulo 3
Revisão Bibliogáfica
Neste capítulo é apresentada uma revisão sobre os assuntos mais relevantes a esse trabalho. Primeiramente é apresentada uma revisão sobre o átomo de carbono e seus diversos alótropos. A seguir, revisamos os nanotubos de carbono e sua morfologia. A seguir apresentamos algumas propriedades dos surfatantes (ou anfifílicos). Por fim, fazemos um breve comentário a respeito do comportamento dos nanotubos de carbono em solução.
3.1 O Carbono e Seus Alótropos
O carbono é um dos elementos mais abundantes do universo, sendo um elemento impressio-nante, sobretudo devido às suas ligações químicas. De acordo com o número de suas ligações químicas ele pode ser divalente (2 ligações), trivalente (3 ligações) ou mesmo tetravalente (4 ligações). Dependendo da natureza da ligação entre dois carbonos adjacentes, diferentes estru-turas podem ser geradas. Na forma sólida o carbono existe, principalmente, em duas estruestru-turas diferentes: o carbono e o grafite, que podem ser considerados duas formas naturais cristalinas de carbono puro. As propriedades e características dessas estruturas são completamente diferentes e podem ser explicadas através das ligações entre os átomos de carbono na estrutura.
3.1. O CARBONO E SEUS ALÓTROPOS
Na forma de grafite, os átomos de carbono se encontram no estado de hibridização sp2. Nesse caso, os átomos de carbono formam uma rede hexagonal plana onde cada átomo está for-temente ligado a três outros átomos vizinhos através de uma ligação do tipoσcom comprimento
1,42 Å. O ângulo entre três átomos de carbono consecutivos é de 120o. O espaçamento entre
as camadas de grafite é de 3,35 Å. A Figura3.1ilustra essa estrutura. As “folhas” de grafite se mantêm unidas através de forças de van der Waals. Porém, a intensidade dessas forças não é suficiente para impedir que as camadas de grafite se desloquem umas sobre as outras quando uma força externa é aplicada, de forma que o grafite se apresenta como um material macio, sendo muito útil em aplicações como lubrificante.
Na forma de diamante os átomos de carbono, se encontram no estado de hibridizaçãosp3. Os átomos formam uma rede tridimensional onde cada átomo de carbono está rodeado tetraedri-camente por 4 carbonos eqüidistantes, conforme a Figura3.2. Estes átomos, por sua vez, ligam entre si através de quatro fortes ligações covalentes, fato este que torna o diamante o polimór-fico mais compacto de carbono, bem como o material de maior densidade e dureza encontrado na natureza. Esta última lhe dá propriedades de corte e polimento de maior importância nas mais diversas áreas industriais. O comprimento de cada uma dessas ligações é de 1,56 Å e a estrutura possui um band gapde 5,5 eV, sendo, portanto, um isolante. Além disso, o diamante possui uma estrutura muito rígida e estável.
Figura 3.2: Célula unitária do diamante. Figura extraída da referência [9].
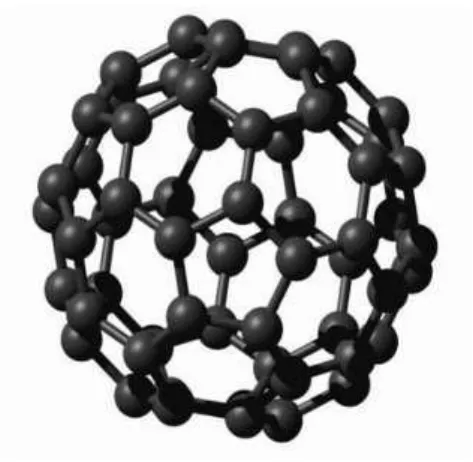
Além dessas duas formas existe ainda o fulereno, descoberto em 1985 por Harold W. Kroto, Robert F. Curl, e Richard E. Smalley, obtido por condensação em uma atmosfera de gás inerte [10]. Por esta descoberta, Kroto, Smaley e Curl obtiveram o prêmio Nobel de Química em 1996. Na estrutura proposta, a molécula de fulereno é formada por 60 átomos de carbono (C60)
que formam uma figura que possui 20 faces hexagonais e 12 faces pentagonais. Essa figura é um domo geodésico oco, chamado icosaedro truncado, cuja aparência se assemelha a uma bola
de futebol com diâmetro de aproximadamente 0,7 nm. Os átomos de carbono noC60 estão em
hibridização intermediária, ou seja, spm onde 2<m<3. Além doC
60, outras estruturas com
3.2. NANOTUBOS DE CARBONO
moléculas quase esféricas de carbono estruturam-se num arranjo cristalino compacto do tipo cúbico de faces centradas com constante de rede de 14,17 Å e a uma distância mínima entre vizinhos de 10,02 Å.
Figura 3.3: FulerenoC60. Figura extraída de http://lqes.iqm.unicamp.br, em 29/04/08.
Outra forma alotrópica do carbono, que possui enorme interesse no meio científico, é o na-notubo de carbono. Este, por sua vez, será descrito nos tópicos a seguir.
3.2 Nanotubos de Carbono
Desde que foram descobertos em 1991 por Iijima [3], usando o processo de pirólise de grafite em plasma sob atmosfera controlada de hélio, os nanotubos de carbono têm sido foco de intenso estudo por pesquisadores de todo o mundo devido às propriedades que possuem e que são de grande interesse nas mais diversas áreas.
Os nanotubos de carbono têm mostrado propriedades excepcionais que sâo conseqüência de sua estrutura altamente simétrica [11]. Muitos pesquisadores têm obtido valores para essas propriedades que excedem os valores para qualquer outro material conhecido. Entre essas pro-priedades se encontram as propro-priedades térmicas, como a condutividade térmica, propro-priedades mecânicas, como rigidez e resistência, e propriedades eletrônicas. Diversos estudos teóricos e experimentais têm mostrado como resultado que os nanotubos de carbono podem apresentar modulo elástico extremamente alto (maior que 1 TPa) e resistência mecânica de 10 a 100 vezes maior que o aço mais forte (em fração de peso) [12]. Ademais, eles são termicamente está-veis sob temperaturas superiores a 2800oC no vácuo e possuem condutividade térmica cerca de
3.2. NANOTUBOS DE CARBONO
a dos fios de cobre. As propriedades eletrônicas dos nanotubos de carbono possuem grande interesse na área da nanotecnologia principalmente na substituição do silício na fabricação de dispositivos diversos, tais como transistores. Devido às suas dimensões seria possível produ-zir dispositivos muito menores e mais rápidos (aproximadamente 1 terahertz ou mais). Além disso, a alta condutividade térmica desse material facilitará a eliminação do calor gerado pelo dispositivo durante seu funcionamento. Já as excepcionais propriedades mecânicas combinadas com a baixa densidade apresentada pelos nanotubos de carbono oferecem grandes possibilida-des de aplicações em compósitos. Pesquisadores têm proposto o uso possibilida-dessas características no desenvolvimento de carcaças de carro e coletes a prova de bala, por exemplo. Ambos seriam super-resistentes e muito mais leves do que os sistemas atuais disponíveis comercialmente.
3.2.1 Morfologia
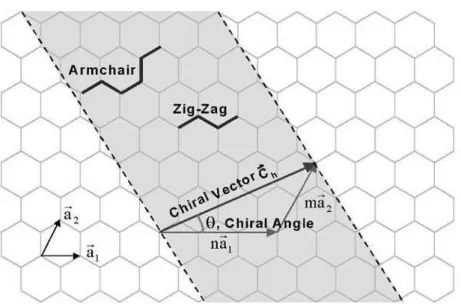
O grafite pode ser visualizado como uma folha 2-D de um arranjo hexagonal de átomos de carbono, que, por sua vez, possuem 3 vizinhos mais próximos. Enrolando essa folha de grafite em cilindros é formado o que se denomina nanotubos de carbono. Dessa forma, cada nanotubo de carbono pode ser visualizado como uma molécula simples devido ao seu pequeno tamanho, ou como cristais quase dimensionais com peridiocidade translacional ao longo do tubo [14].
A estrutura atômica dos nanotubos é descrita em termos da quiralidade dos seus tubos (ou heliodicidade) que é definida pelo vetor quiral C~h, e ângulo quiral, θ. Na Figura 3.4 é
mos-trado uma folha de grafite com os vetores unitários da rede ~a1 ea~2, vetor quiralC~h (ou vetor
de rolamento) e ângulo quiralθ. O vetor quiral pode ser expresso em função dos vetoresa~1ea~2:
~
Ch=n~a1+m~a2 (3.1)
onde os inteiros ne m são os números de passos ao longo das ligações “zig-zag” do carbono
da rede hexagonal. Os pontos ligados pelo vetor quiral são cristalograficamente equivalentes e o nanotubo é obtido pelo dobramento do plano até que estes pontos se tornem coincidentes. Logo, o diâmetro dos tubos é definido pelo móduloChdo vetor quiral, com base no tamanho do espaçamento interatômico entre os átomos de carbono:
Ch=M
µ n M|~a1|+
m M|a~2|
¶
(3.2)
ondeM é o maior divisor comum entrenem. A direção deCh é medida pelo ângulo quiralθ,
3.2. NANOTUBOS DE CARBONO
cosθ= ~a1·C~h |~a1||C~h|
Figura 3.4: Representação de uma folha de grafite. Os vetoresa1ea2 são os vetores unitários
da rede hexagonal. Figura extraída da referência [11].
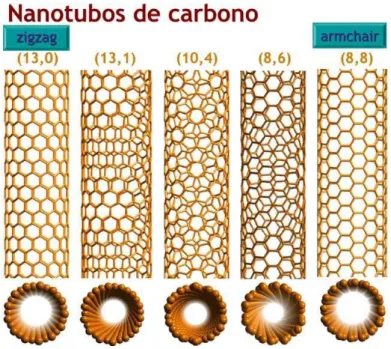
θdefine a quantidade de “giro” que o tubo apresenta. Há dois casos limites para o valor deθ: 0o
e 30o. No primeiro caso, quandoθ=0o, o nanotubo possui índices do tipo(n,0)e é denominado
do tipo zig-zag por exibir um padrão de “zigue-zague” ao longo da circunferência do tubo.
No segundo caso, quando θ =30o, o nanotubo possui índices do tipo (n,n) e é denominado
armchair. Os demais casos, onde 0<θ<30o, os nanotubos são chamamos quirais. Na estrutura
armchair duas ligações C-C em lados opostos de cada hexágono são perpendiculares ao eixo do nanotubo enquanto na configuraçãozig-zagestas ligações são paralelas. Nos tubos do tipo quiral as ligações C-C estão formando um ângulo com o eixo do nanotubo. A Figura3.5ilustra estes três tipos de nanotubos.
O diâmetro de um nanotubo é obtido através do comprimento do vertor quiral:
d= |C~h|
π =
a0
π p
n2+2n m+m2= a0 π
√
N (3.3)
3.2. NANOTUBOS DE CARBONO
Figura 3.5: Tipos de nanotubos segundo a quiralidade. Os pares de números acima de cada tubo indicam os índices correspondentes a cada um desses tubos. Figura extraída de http://cienciasnaedem.blogspot.com, em 29/04/08.
~a=−2m+n
M R a~1+
2n+m M R ~a2
(3.4)
e
|~a|=a=
p
3(n2+n m+m2)
M R a0 (3.5)
onde
R=
(
3 , se n−m
3M é um número inteiro
1 , nos demais casos
3.2. NANOTUBOS DE CARBONO
aZ =
√
3a0 (3.6)
|C~Z|=M a0 (3.7)
para ozig-zage:
aA=a0 (3.8)
|C~A|=
√
3M a0 (3.9)
Tubos de mesma quiralidade possuem o mesmo vetor~a. O valor do módulo de~a varia forte-mente com a quiralidade dos tubos. Tubos quirais freqüenteforte-mente têm células unitária muito compridas. A Figura3.6nos mostra alguns tipos de nanotubos destacando sua célula unitária.
Figura 3.6: Comprimento da célula unitária segundo a quiralidade. Note que tubos quirais apresentam células unitárias mais compridas, o que significa maior período translacional. Fi-gura extraída da referência [15].
Podemos calcular o número de hexágonos da célula unitária de um nanotubo a partir da razão entre a área da superfície do cilindro Sc =a ce da área da célula unitária da rede do grafeno
(Sgra f):
Nhex=
Sc
Sgra f
= 2((n
2+n m+n2))
M R (3.10)
3.2. NANOTUBOS DE CARBONO
nc=2.Nhex=
4((n2+n m+m2))
M R (3.11)
Para os tuboszig-zagearmchair nc=4n.
3.2.2 Nanotubos de carbono de paredes múltiplas
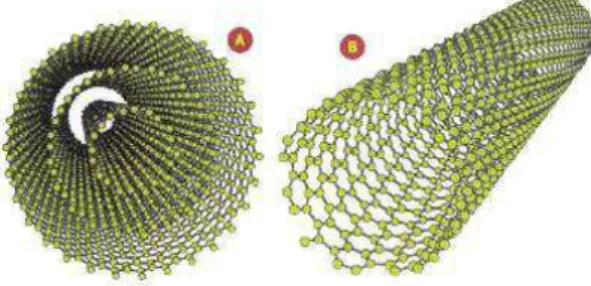
Os nanotubos de carbono podem ser divididos em duas categorias bem distintas: nanotubos
de carbono de parede simples (SWNT - single wall nanotubes), constituídos por apenas uma
camada cilíndrica de grafite, e nanotubos de carbono de paredes mútiplas (MWNT -multi wall nanotubes), constituídos de vários cilindros concêntricos de grafite.
Os SWNT apresentam características físicas de sólidos, podendo ser considerados como cristais. Eles podem ser fechados em seus extremos com hemisférios de fulerenos. Obser-vações experimentais indicam que os dimâetros dos SWNT variam entre 0,7nm e 3nm [16]. Alguns pesquisadores conseguiram produzir SWNT com comprimentos admirável, em torno de 4cm [17].
Figura 3.7: Nanotubos de parede simples (à direita) e de paredes múltiplas (à esquerda). Ima-gem extraída de http://ramonflores.br.tripod.com, em 29/04/08.
3.3. SURFACTANTES
Muitas da propriedades dos MWNT são bem próximas as do grafite. Suas propriedades eletônicas, por sua vez, não são tão bem definidas devido a possibilidade de um grande número possível de camadas.
Do ponto de vista das investigações teóricas, os SWNT representam os sistemas mais ade-quados devido à maior facilidade de descrição computacional. Porém os MWNT são produzidos com mais facilidade, com menor custo e possuem uma vasta gama de possíveis aplicações [19]. As propriedades dos nanotubos de carbono dependem do arranjo atômico, do diâmetro, comprimento e da morfologia dos tubos. A quiralidade, por exemplo, possui um grande im-pacto nas propriedades eletrônicas do material. O grafite é considerado um semi-metal. Porém, de acordo com a quiralidade, os nanotubos podem ser metálicos ou semicondutores. Estudos demonstram que a quiralidade também influencia nas propriedades mecânicas dos nanotubos [20].
3.3 Surfactantes
Os surfactantes (ou agente tensoativos) são compostos que possuem atividade na superfície da interface entre duas fases como água e óleo ou água ar, por exemplo. Eles se caracterizam por possuir duas regiões distintas na mesma molécula: uma região que é polar, hidrofílica, e outra região não-polar, hidrofóbica. Por isso, comumente, as moléculas que possuem propriedades surfactantes são chamadas moléculas anfifílicas. Essa particularidade na estrutura química dos surfactantes é responsável pelos fenômenos de atividade na tensão superficial de interfaces, pela formação das micelas (micelização) e solubilização [21].
Uma das características dos surfactantes é a capacidade de formar agregados em solução a partir de uma determinada concentração, chamada concentração micelar crítica (cmc). A cmc é uma propriedade intrínseca dos surfactantes. A partir dessa concentração ocorre a formação de agregados termodinamicamente estáveis e facilmente reprodutíveis [22]. Entre esses agregados estão as micelas, que são estruturas organizadas mais ou menos esféricas, de dimensões coloi-dais (entre 3 a 6nm [23]), o que representa uma quantidade em torno de 30 a 200 moléculas
do surfactante (ou monômeros). Na Figura 3.8 está representado uma micela bidimensional
formada por anfifílicos de cauda simples.
Existem, além das micelas, outros agregados termodinamicamente estáveis que podem ser formados por moléculas anfifílicas em sulução a uma concentração maior que a cmc, como vesículas e lamelas. Essas configurações são mais comuns em sistemas formados por anfifílicos de cauda dupla, mas podem ser obtidos em sistemas onde estão presentes apenas anfifílicos de cauda simples [24]. Na Figura3.9há uma representação de uma visícula unilamelar.
3.3. SURFACTANTES
Figura 3.8: Representação de uma micela. Figura extraída da ref [23]
Figura 3.9: Representação esquemática de uma lamela.
sutis de capacidade calorífica e volume que acompanham o processo de micelização.
As micelas são responsáveis pela grande maioria das aplicações práticas dos surfactantes nas mais diversas áreas devido à sua importante propriedade de promover a solubilização de compostos pouco solúveis no meio em que se encontram. Quando em soluções aquosas, os surfactantes alteram muitas propriedades físico-químicas da água, como a sua tensão superficial, viscosidade e condutividade elétrica, por exemplo. Os métodos usuais para a determinação da cmc de um surfactante qualquer acompanham as mudanças em alguma propriedade físico-química da solução em função da concentração de surfactante, detectando a região onde ocorre uma mudança acentuada na variação dessa propriedade. O valor da concentração do surfactante nesse ponto é a cmc do surfactante, ou seja, a partir desse valor ocorre a micelização. A Figura 3.10ilustra essa característica.
3.4. COMPORTAMENTO EM SOLUÇÃO AQUOSA
Figura 3.10: Variação da tensão superficial da água em função da concentração de moléculas detergentes.
3.3.1 Dodecil Sulfato de Sódio
O dodecil sulfato de sódio, ou SDS, é um anfifílico aniônico de cauda simples que possui a fórmulaCH3(CH2)10−CH2−O−(SO3)−−Na+. Sua molécula é constituída de uma cadeia alquílica com um ramo de 12 átomos de carbono, praticamente insolúvel em água, ligada co-valentemente a um grupo iônico, o sulfato de sódio, dando à molécula propriedades anfifílicas requeridas por um detergente (ver Figura3.11).
Figura 3.11: Esquemas representativos da molécula do SDS em 2D (acima) e 3D (abaixo). Figura extraída de http://en.wikipedia.org, em 29/04/08.
O diâmetro da micela varia com o tamanho da cauda da molécula. Para o SDS esse valor é aproximadamente 5,6 nm (5,0±1 a 6,3±2 nm) e não varia com a sua concentração [25].
O SDS possui grande interesse na área da simulação computacional devido à simplicidade apresentada por essa molécula.
3.4 Comportamento em Solução Aquosa
3.4. COMPORTAMENTO EM SOLUÇÃO AQUOSA
descritos como partículas em forma de bastões e o comportamento de objetos rígidos delgados em líquidos tem sido estudado ha décadas.
O maior obstáculo encontrado no desenvolvimento de aplicações dos nanotubos de carbono tem sido a diversidade dos diâmetros e da quiralidade dos tubos e os estados de agregação nas amostras obtidas nos vários métodos de preparação. Isso, aliado com a dificuldade de dispersão dos nanotubos em líquidos e à falta de técnicas de observação da dinâmica destes em solução têm tornado lento o progresso fundamental do entendimento do comportamento dos nanotubos em solução em fase líquida [26]. Esse comportamento é de suma importância na física, na ciência de materiais e nas ciências da vida. A agregação é um grande problema porque os nanotubos são altamente polarizáveis e tendem a formar feixes de cordas paralelas que se atraem por forças de van der Waals com uma energia média de ligação de aproximadamente 500 eV por micrômetro de contato entre os tubos [6]. Isto perturba a estrutura eletrônica dos tubos e frustra as tentativas de separar os tubos por tamanho, tipo ou quiralidade e usá-los como espécies macromoleculares. Um procedimento proposto que visa resolver esse problema seria a aplicação de um método que cause desagregação dos nanotubos dos feixes por meios físicos aplicando um revestimento em torno destes para prevenir que eles se agreguem novamente, e então remover os feixes restantes da solução.
Um método utilizado para se separar os nanotubos dos feixes é a modificação covalente destes com o uso de polímeros ou compostos orgânicos de baixo peso molecular. Porém as aproximações produzem ligações de materiais sobre os nanotubos, destruindo sua rede contínua de ligações eletrônicas do tipoπ. Consequentemente, isso resulta na perda de suas propriedades
eletrônicas. Outra forma que tem sido estudada para se obter a separação dos nanotubos é atra-vés da adsorção de moléculas. Essa técnica de solubilização mantém intacta a estrutura perfeita dos nanotubos de carbono e aumenta a sua dispersão em uma ampla variedade de solventes polares, não polares e matrizes poliméricas [27].
Diversos trabalhos já foram realizados utilizando SDS adsorvidos na superfície de nanotu-bos de carbono. No trabalho realizado por O’Connell e colaboradores [6], nanotunanotu-bos de carbono foram colocados em solução aquosa na presença de SDS, que foram adsorvidos em sua super-fície formando micelas cilíndricas. Através de agitação ultra-sônica e centrifugação, obtiveram uma suspensão rica em SWNT. Além disso, também foi simulada a adsorção de SDS por nano-tubos de carbono através de dinâmica molecular em temperatura e pressão constantes e, através desse método, constataram que a cauda hidrofóbica das moléculas de SDS pode adotar diversas orientações diferentes de acordo com o tubo. Além disso, existe probabilidade de que as molé-culas de SDS se entortem ou girem de forma que os átomos de carbono atinjam a posição média radial dos átomos de enxofre. As moléculas de água apresentam certa probabilidade de penetrar entre as caudas hidrofóbicas do anfifílico. Isso foi observado nas simulações de micelas cilín-dricas puras de SDS. Porém, não foram observado moléculas de água nas vizinhanças do tubo, indicando que os tubos individuais estão em um meio formado apenas de hidrocarbonetos.
Pas-3.4. COMPORTAMENTO EM SOLUÇÃO AQUOSA
quali [26]. Em seu trabalho eles descreveram uma forma de se visualizar nanotubos de carbono individuais em suspensão aquosa em tempo real através do uso de um corante hidrofóbico flu-orescente. O corante é adicionado na solução contendo os nanotubos de carbono encapsuladas por SDS, formando uma micela alongada. Devido à sua hidrofobicidade, o corante se incorpora espontaneamente no corpo das micelas de SDS. Dessa forma o comportamento dos nanotubos de carbono em suspensão aquosa pode ser visualizado através de microscopia de fluorescência. Outro trabalho de grande interesse foi realizado por Bandyopadhyaya et al [28]. Em seu trabalho, promoveram a solubilização de nanotubos em solução aquosa na presença de um polímero natural de cadeia longa conhecido como goma arábica. A goma arábica foi adsorvida, promovendo a separação dos tubos e consequúente solubilização sem danificar os tubos nem alterar suas propriedades. Após a secagem, os nanotubos obtidos através desse processo estão prontos para o uso, não necessitando de nenhum outro processamento.
Capítulo 4
Metodologia
Neste capítulo apresentamos uma breve descrição do Método de Monte Carlo, comentando suas principais características e relacionando as mesmas com os princípios da Mecânica Estatística. Ao final, apresentamos também uma breve descrição da função de estrutura e seu papel na análise de nossos resultados.
4.1 O Método de Monte Carlo
OMétodo de Monte Carloé um poderoso método de simulação, destinado, principalmente, ao estudo das propriedades de sistemas em estado de equilíbrio termodinâmico, através do uso de conceitos e princípios da mecânica estatística de equilíbrio.
A mecânica estatística é o ramo da física que trata do cálculo das grandezas termodinâ-micas que caracterizam um sistema de muitas partículas, através das médias dessas grandezas sobre intervalos de tempo e regiões espaciais comparáveis aos da resolução experimental [29].
Imagine que queiramos descrever o comportamento dinâmico de um sistema comN partíulas
microscópicas, onde N≈1023 partículas. Para isso, precisamos conhecer como variam a
po-sição e a velocidade de cada partícula em função do tempo. Logo, teríamos um sistema com
N equações acopladas, cada uma descrevendo o comportamento de uma partícula. Além disso,
seria necessário aplicar condições iniciais suficientes para se resolver esse sistema, o que seria impossível. Em casos como esse, onde não é possível se obter uma solução exata, utilizamos outros métodos para se estudar o problema e obter uma solução.
O algorítmo de Monte Carlo é um exemplo de um desses métodos. Partindo da hamilto-niana do modelo físico que se deseja estudar, geramos configurações aleatórias do sistema, de acordo com a distribuição de probabilidades com peso de Boltzmann adequado e normalizado, até encontrarmos uma configuração de equilíbrio. Dessa forma, as médias de variáveis termo-dinâmicas se tornam médias simples.
Considerando um sistema em contato com um reservatório térmico cuja temperatura éT,
4.1. O MÉTODO DE MONTE CARLO
Boltzmann:
Pieq=e− βEi
Z (4.1)
ondeβ= k1
BT,kbé a constante de Boltzmann,Eié a energia do estadoieZé a função partição, que é definida como:
Z=
∑
i
e−βEi (4.2)
A soma em (4.2) é realizada sobre todos os estados do sistema. A função partição possui ex-trema importância na mecânica estatística, visto que a partir dela pode-se obter diversas
gran-dezas termodinâmicas de interesse. O valor esperado de qualquer grandeza A de um sistema
cujos graus de liberdade sejam discretos, pode ser obtida através da média em todos os estados do sistema com o peso dado pela distribuição de Boltzmann:
hAi= 1
Z
∑
i Aie−βEi (4.3)
ondehAirepresenta a média deAno ensemble, ou seja,hAié a média aritmética dos valores de Asobre todos os sistemas do ensemble. Caso o sistema apresente graus de liberdade contínuos, a soma deve ser substituída por uma integral.
Porém, a soma em (4.2) só é possível ser realizada sobre todos os estados para sistemas muito pequenos ou muito simples. Para o estudo de sistemas reais, uma solução é obter a média apenas num subconjunto de estados, mesmo que, dessa forma, introduzamos necessariamente um erro ao cálculo.
A média no ensemble (4.3) é aproximada pela soma em um subconjunto deM estados
to-mados com probabilidadePi, ondeienumera os estados:
hAi ≈A= ∑
M
i AiPi−1eβEi
∑Mi Pi−1eβEi (4.4)
À medida queMcresce tem-se uma estimativa cada vez melhor dehAi. Uma importante ques-tão é como escolher os estadoside forma que se obtenha a melhor estimativa parahAi. Caso as
4.1. O MÉTODO DE MONTE CARLO
uma amostragem representativa desse espaço se considerarmos um número muito grande de estados. Isso seria proibitivo em termos do tempo computacional que seria dispendido nesse cálculo. Uma forma de se otimizar o cálculo dehAiseria escolher com maior probabilidade os estados mais significativos. A esse tipo de amostragem denominamos amostragem por impor-tância. Uma escolha simples paraPi é a distribuição de Boltzmann e o cálculo dehAise torna
uma média aritmética:
A= ∑
M i Ai
M (4.5)
4.1.1 Processos Estocásticos
Definimos como Processo Estocástico uma coleção de variáveis aleatórias X(t)indexadas por
um parâmetrot pertencente a um conjuntoT ondeX(t)representa uma característica
mensu-rável de interesse em um instante de tempot. Os processos estocásticos são de interesse para descrever o procedimento de um sistema operando sobre algum período de tempo. Assim, a variável randômicaX(t)representa o estado do sistema no parâmetrot(geralmentetrepresenta o tempo). Logo,X(t)é definido em um espaço denominadoEspaço de Estados.
Seja um processo estocástico que obedeça a seguinte relação:
P{X(tk+1) =xk+1|X(tk) =xk,X(tk−1) =xk−1, ...,X(t1) =x1,X(t0) =x0}=
=P{X(tk+1) =xk+1|X(tk) =xh} (4.6)
Nesse caso, esse processo é dito ser um Processo Markoviano. De uma forma mais simples:
4.1. O MÉTODO DE MONTE CARLO
4.1.2 Cadeias de Markov
Umacadeia de Markové todo processo markoviano cujas variáveis aleatóriasX(t)estão
defini-das em um espaço de estados discretos. Quando o tempo é discreto, a cadeia de Markov é dita ser uma cadeia de Markov de tempo discreto. A probabilidade de transição entre um estadoSi
para um estadoSj qualquer é dada por:
P(Si→Sj) =P(Xtn=Sj|Xtn−1 =Si) (4.7)
Essas probabilidades devem satisfazer as seguintes condições:
• Positividade: P(xi→xj)≥0 , para qualquer que sejamie j.
• Conservação ou normalização:∑jP(xi→xj) =1
Outras condições que a cadeia de Markov deve obedecer são a condição da ergodicidade e a condição do balanço detalhado.
Ergodicidade: Em um processo Markoviano, qualquer estadoxipode ser gerado a partir de
ou-tro estadoxjdesde quetseja suficientemente grande. Isso vem do fato de que cada estado
tem uma probabilidade diferente de zero de acordo com a distribuição de Boltzmann.
Balanço detalhado: SejaP(xi,tn)a probabilidade de encontrarmos o sistema em xi no tempo
t=tn. A evolução deP(xi,tn)é descrita pela equação mestra
dP(Sj,t)
dt =
∑
i P(Si→Sj)P(Si,t)−∑
i P(Sj→Si)P(Sj,t) (4.8) O primeiro termo do lado direito dessa expressão descreve a soma da taxa de todas as transições de todo o espaço de fases para a configuraçãoxjem um dato instante de tempot, enquanto o segundo termo da expressão descreve a soma das taxas de transições para a configuraçãoxj. Quando em equilíbrio (regime estacionário) as taxas em que o sistema
realiza transições para o estado je fora dele devem ser iguais. Logo:
dP(Sj,t)
4.1. O MÉTODO DE MONTE CARLO
Da condição da conservação temos:
∑
i
P(Sj→Si)P(Sj,t) =P(Sj)
∑
iP(Sj→Si) =P(Sj) (4.10)
que implica em:
P(Sj) =
∑
iP(Si)P(Si→Sj) (4.11)
que é a condição de balanço ou estacionaridade. Como dP(Sj,t)
dt =0 qualquer conjunto
P(xi→xj)que satisfaça a equação (4.10) tenderá para uma distribuição de equilíbrio.
A partir daqui podemos definir uma condição mais abrangente que o balanço detalhado no regime estacionário: a condição de balanço detalhado, que leva a conversão de P a partir de qualquer estado inicial do processo markoviano:
P(Sj→Si)P(Sj,t) =P(Si→Sj)P(Si,t) (4.12)
Através da equação (4.2), dado que a distribuição de equilíbrio é a distribuição de Boltzmann, temos:
P(Si→Sj)
P(Sj→Si)
=P(Sj)
P(Si)
=e−β(Ej−Ei) (4.13)
Para que um conjunto de probabilidades qualquer possa ser usado em simulações de Monte Carlo é necessário que ele satisfaça a equação acima. Um exemplo de um algoritmo que utiliza conjunto de probabilidades desse tipo é o famosoalgorítmo de Metrópolis.
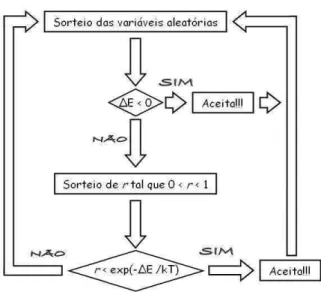
4.1.3 O Algoritmo de Metrópolis
4.2. FUNÇÃO DE ESTRUTURA
um estadoxipara um outro estadoxj são dadas por:
P(Si→Sj) =
(
e−β(Ei−Ej) , se∆E>0
1 , se∆E≤0
onde∆E =Ei−Ej. Essa relação estabelece as regras de aceitação ou rejeição de alguma
mu-dança realizada na configuração do sistema. Ela caracteriza o algorítmo de Metrópolis. Supo-nhamos que em um instantet qualquer o sistema esteja em estadoxi, cuja energia sejaEi. O
procedimento de sorteio das transições é feito da seguinte forma:
1) Sorteamos um novo estado xj no instante de tempo t+1, numa forma que seja
apropri-ada ao modelo do sistema físico estudado; 2) Calculamos a energiaEj do estadoxj;
3) CasoEj≤Eia nova configuração é aceita e a simulação procede para o passo 5.
4) CasoEj>Eientão sorteamos um númerortal que 0<r<1. Ser≤e−β(Ei−Ej)a nova
con-figuração é aceita e sorteamos uma nova concon-figuração a partir dela. Caso contrário, rejeita-se essa configuração e retornamos ao item 1.
5) Calcula-se os valores das macrovariáveis de interesse. 6) Retorna ao passo 1 em um instante de tempo posterior.
Após um número suficientemente grande de sorteios (“tempo” suficientemente grande) o sistema atingirá a configuração de equilíbrio e os valores de equilíbrio podem ser obtidos através da equação (4.5).
Vale lembrar que em simulações de Monte Carlo de diversos modelos físicos, o tempo não tem o mesmo sentido do tempo real, pois não há equações de movimento temporal, somente uma estatística sobre a sequência de configurações geradas com probabilidade de Boltzmann.
Logo,t serve apenas como o contador do número de configurações geradas. Porém, quando o
movimento das partículas se dá através de movimentos locais há uma relação entre o tempo em Monte Carlo e o tempo em dinâmica molecular, mesmo na ausência destas equações ([31]).
A Figura4.1esquematiza esse algoritmo.
4.2 Função de estrutura
No intuito de se obter a organização da estrutura local da configuração de equilíbrio, faremos uso da função de distribuição radial, g(r). Essa função está relacionada com a probabilidade
4.2. FUNÇÃO DE ESTRUTURA
Figura 4.1: Algoritmo de Metropolis.
ρg(r) = 1
N N
∑
i N∑
j6=i
δ[~r−~ri j] (4.14)
ondeN é o número total de átomos,ρ= NV é a densidade,ri j é a distância entre os centros de
dois átomos i e j e δ está relacionado com a posição do centro do átomo, podendo assumir
valores entre[o,∞]. A normalização deg(r)é obtida a partir da expressão:
ρg(r) = 2
Nh N
∑
i N∑
j<i
Z
δ[~r−~ri j]d~ri (4.15)
A integral acima é realizada sobre todas as separações possíveis entre dois átomos.
A funçãog(r)pode ser escrita de forma probabilística explicitamente para todos osM pas-sos da simulação:
g(r) = ∑
M
k=1Nk(~r,∆~r)
M¡12N¢ρV(~r,∆~r) (4.16)
onde∆~ré a espessura de uma camada fina e finita,Nk(~r,∆~r)é o número de átomos encontrados
4.2. FUNÇÃO DE ESTRUTURA
O fator de estrutura é o mesmo medido em experimentos de espalhamentos de nêutrons e de raios-X. Ele é obtido através da transformada de Fourier de g(r)[33, 34, 35]. O estudo da
função g(r) nos permite determinar a distância média dos vizinhos através do valor máximo
Capítulo 5
Modelo
Soluções que apresentam moléculas surfactantes são normalmente enquadradas na classe dos fluídos complexos. Tipicamente, essas moléculas possuem diferentes tipos de átomos, dife-rentes tipos de interações entre os constituintes e um grande número de graus de liberdade internos, tornando difícil a simulação de um modelo atomístico detalhado. Além disso, a carac-terística dos surfactantes de formarem estruturas supramoleculares, como as micelas, dificulta a obtenção do equilíbrio térmico do sistema [36]. Ademais, simulações de sistemas de muitas partículas onde as moléculas de água aparecem explícitamente são mais demoradas, pois estas dificultam ainda mais a termalização, devido ao grande número de movimentos realizados em cada passo. Devido a esses e outros fatores, o estudo de um modelo minuciosamente detalhado demanda um grande tempo computacional para as simulações, o que é, na maioria das vezes, um problema. Assim, simplificações no modelo adotado se tornam necessárias.
Figura 5.1: Figura ilustrativa de um modelo de partículas em uma caixa com posições discreti-zadas (à esquerda) e contínua (à direita).
5.1. MOLÉCULAS E MOVIMENTOS
dentro de uma caixa de paredes fixas (sistemas “livres de rede”). No primeiro caso, a interação entre as partículas acontece de forma mais simples, visto que não se torna necessário o cálculo da distância entre as partículas a cada movimento; as distâncias são definidas pelas posições na rede, simplificando o cálculo da energia do sistema. No caso da ausência de rede, a cada novo movimento a partícula pode assumir infinitas posições no espaço, necessitando o recálculo da distância entre as partículas para se encontrar a nova energia.
Nesse trabalho, desenvolvemos um modelo livre de rede com partículas de água explícitas. Todas as simulações são feitas com partículas em uma caixa bidimensional quadrada de paredes rígidas de tamanhoLem uma temperatura fixaT (ensemble canônico). A representação de cada objeto do modelo (moléculas surfactantes, moléculas de água ou nanotubos de carbono) está detalhado nos tópicos a seguir.
Uma importante questão a ser definida é como serão realizados os movimentos de cada mo-lécula. A importância dessa escolha está no fato de que a condição da ergodicidade deve ser respeitada (Sessão4.1.2). A escolha dos possíveis movimentos também irá ditar o quão rápido nosso modelo evolui para a configuração de equilíbrio. Os movimentos das moléculas devem permitir que o sistema atinja todas as configurações possíveis em um intervalo suficientemente grande de tempo. A idéia inicial é, ao sortear um objeto no espaço, escolhemos aleatoriamente uma das formas pré-definidas de se mover esse objeto e realizamos a tentativa de se obter a nova configuração. Os detalhes dos movimentos escolhidos para cada objeto são descritos a seguir.
5.1 Moléculas e Movimentos
Nessa seção é definido detalhadamente cada molécula e os respectivos movimentos adotados para cada uma.
5.1.1 Molécula de Água (
w
)
Por simplificaão, as moléculas de água serão representadas por uma partícula apenas. Devido à distância de exclusão nos potenciais adotados, cada partícula do sistema se comporta como um disco rígido no espaço. Devido à simplicidade, as moléculas de água só se movem por transla-ções no espaço, cujas distâncias de deslocamento são definidas por:
δwx =εx∆w (5.1)
5.1. MOLÉCULAS E MOVIMENTOS
ondeδwx eδwy são as distâncias de deslocamento da translação na direçãoxey, respectivamente, εxeεysão números aleatórios obtidos de uma distribuição uniforme entre 0 e 1 e∆wé a
distân-cia máxima de deslocamento para as moléculas de água.
5.1.2 Molécula Anfifílica (
surf
)
Em dinâmica molecular, cada par de átomos ligados possui energia vibracional que faz com que eles vibrem como osciladores. Dessa forma a distância de ligação entre os átomos varia no decorrer do tempo. Além disso, uma molécula pode apresentar ainda energia rotacional e variação no ângulo da ligação, por exemplo, o que gera mais graus de liberdade no sistema. Porém, no modelo desenvolvido nesse trabalho, as moléculas anfifílicas são moléculas que, apesar de possuírem certa flexíbilidade, apresentam ligações de tamanhos e ângulos fixos. Logo, o efeito da oscilação, rotação ou qualquer tensão sobre as ligações é desconsiderado.
Os ângulos das ligações entre os átomos de carbono só poderão assumir um valor fixo, correspondente ao ângulo de ligação da ordenação tetraédrica do carbono. O valor desse ângulo é de 109,5oentre três átomos subseqüentes (duas ligações). A cabeça da molécula anfifílica (h)
será designada por um ponto no espaço. Cada átomo da cauda carbônica (t) será representado por pontos subseqüentes a este. No caso da molécula do SDS, essa cauda possui 12 pontos. Em nosso modelo adotamos uma molécula anfifílica com uma partícula na cabeça e 4 partículas na cauda (h1t4).
Nesse trabalho adotamos 4 possíveis movimentos para a molécula anfifílica: o movimento de reptação, de translação, de rotação e o movimento de pivô. Cada um desses movimentos é descrito a seguir.
5.1.2.1 Reptação
5.1. MOLÉCULAS E MOVIMENTOS
Figura 5.2: Esquema ilustrativo do movimento de reptação. Nesse exemplo a cabeça da mo-lécula surfactante foi sorteada. As setas representam as direções possíveis para se realizar o movimento (a mais clara indica a direção “não escolhida”).
5.1.2.2 Translação
Para que todas as posições possíveis no espaço para a molécula surfactante possam ser acessí-veis, o movimento de translação da molécula se torna necessário. Esse movimento consiste em “arrastar” a molécula selecionada como um bastão rígido em uma quantidadeδsur fx na direção
horizontal eδsur fy na direção vertical de forma que cada ponto que define a molécula se desloque
de uma mesma distânciaδsur f. Os valores paraδsur f
x eδsur fy são obtidos através da relação:
δsur fx =εx∆sur f (5.3)
δsur fy =εy∆sur f (5.4)
ondeεxeεysão dois números aleatórios entre 0 e 1 e∆sur f é o deslocamento máximo permitido
na translação da molécula anfifílica. Dessa forma, há probabilidade de se encontrar a molécula em qualquer posição possível do espaço. A Figura (5.3) ilustra esse movimento.
Figura 5.3: Esquema ilustrativo do movimento de translação.
5.1.2.3 Rotação
5.1. MOLÉCULAS E MOVIMENTOS
5.4) de um ânguloθsur f definido como:
θsur f =εθΘsur f (5.5)
ondeεθ é um número aleatório entre 0 e 1 eΘsur f é o ângulo correspondente ao giro máximo
permitido para cada rotação de uma molécula anfifílica. O ponto fixo no qual ocorrerá a rotação será um átomo qualquer desta molécula sorteado aleatóriamente cada vez que o movimento de rotação do surfactante for realizado. Assim, podemos encontrar qualquer configuração de uma molécula em qualquer “ângulo” possível.
Figura 5.4: Esquema ilustrativo do movimento de rotação. O ponto no centro da partícula indica a posição do eixo de rotação.
5.1.2.4 Pivô
O quarto movimento permitido às moléculas surfactantes será o movimento conhecido como pivô. Nesse movimento, uma ligação da molécula é escolhido de forma aleatória e ela é “do-brada” nessa ligaçã de forma que ela adquira uma nova configuração. Essa dobra ocorre de forma que o ângulo assuma o outro valor possível, respeitando o ângulo de ligação. A Figura 5.5Ilustra esse movimento.
5.1. MOLÉCULAS E MOVIMENTOS
5.1.3 Nanotubo de Carbono (
nc
)
Como já mencionado, os nanotubos de carbono, quando em solução, se agregam formando feixes paralelos de tubos. Em nossas simulações adotamos que todos os nanotubos cortam perpendicularmente o plano estudado. Assim, cada nanotubo será representado por um conjunto de pontos no plano equidistantes do eixo central do nanotubo. Devido a isso, foram simulados
apenas nanotubos de carbono do tipo zig-zag, por apresentarem um número significativo de
átomos de carbono neste plano além de serem igualmente espaçados, o que não ocorre com os nanotubos do tipoarmchair. Cada átomo se distancia do seu vizinho coplanar de uma distância a0. A Figura5.6ilustra esse fato.
Figura 5.6: Número de pontos em um mesmo plano para três tipos de nanotubos. Note que
ematemos representado um nanotuboarmchair, que possui 12 átomos no plano de corte, um
nanotubozig-zagem b, com 8 átomos e em cum nanotubo quiral, com apenas 1 átomo neste
plano. Figura extraída da referência [15] (adaptada).
5.2. HAMILTONIANA
5.1.3.1 Translação
Assim como na molécula surfactante, o movimento de translação do nanotubo de carbono
ocorre de forma que todos os pontos que o definem se desloquem de uma distância δncx na
direçãoxeδncy na direçãoy, definidos da seguinte forma:
δncx =εx∆nc (5.6)
δncy =εy∆nc (5.7)
ondeεx eεy são dois números aleatórios sorteados de uma distribuição uniforme entre 0 e 1 e
∆ncé a distância máxima percorrida por um nanotubo em um passo.
5.1.3.2 Rotação
O movimento de rotação ocorre de forma que todos os pontos que definem o nanotubo girem em torno de um eixo que passa no centro da circunferência que tangencia os pontos do nanotubo de um ânguloΘncdefinido como:
θnc=εθΘnc (5.8)
onde εθ é um número aleatório sorteado de uma distribuição uniforme entre 0 e 1 e Θnc é o
ângulo correspondente ao giro máximo permitido para cada rotação do nanotubo.
5.2 Hamiltoniana
A energia do sistema é calculada através do potencial de interação entre cada partícula com as demais. Dessa forma, a hamiltoniana do modelo é descrito como:
H=−
∑
i,j(i6=j)
Ji j(r)SiSj (5.9)
ondeJi j(r)é a intensidade do potencial entre um par de partículas quaisquer. Si eSj
5.2. HAMILTONIANA
de partículasi,j do sistema. A excessão ocorre para as partículas de uma mesma molécula, o que é o caso de uma molécula surfactante ou de um nanotubo. A energia conformacional para os surfactantes também é desconsiderada nesse modelo. Assim:
Ji j(r) =
(
0 , seie jpertencem a mesma molécula
Ji j(r) , demais casos.
Os potenciaisJi j(r)descrevem a atração ou repulsão entre partículas, onde os índices indicam
um par qualquer de partículas do sistema que interagem. No caso de um par de partículas que se atraem o potencial adotado nesse modelo é uma simplificação do potencial de Lennard-Jones. A forma do potencial está representado na Figura 5.7B. Note que há uma distância de corte igual a 3d, que é a distância máxima que duas partículas interagem, e as moléculas não podem se aproximar de uma distância menor qued, que é o diâmetro de uma partícula. De acordo com esse potencial, duas partículas fazem “ligação” quando a distância ri j entre seus centros está
entre d e 2d. No caso de partículas que se repelem, o potencial adotado possui uma barreira
no lugar de um poço, como na Figura 5.7C. A excessão é o potencial para o par água-água,
que possui a forma da Figura5.7A. Esse potencial possui uma distância de exclusão, limitando
a distância mínima permitida entre duas partículas em d. A seguir, possui uma barreira de
potencial de energia 6E0 para valores de rww entre 2d e 3d. As partículas de água se ligam
quando 2d≤rww≤3d com uma energia deE0. Para distâncias maiores que 3d as partículas
não interagem.
A energia de interação entre cada par possível de partículas está descrita na Tabela5.1. Ex-ceto pela repulsão entre as águas, todos os demais potenciais são, por simplificação, adotados como sendoE0=1.
w h t nc
w +1 +1 -1 -1
h +1 +1 -1 -1
t -1 -1 +1 +1
nc -1 -1 +1 +1
Tabela 5.1: Possíveis valores paraE0. A intensidade da repulsão entre duas moléculas quaisquer
comri j≤dé∞(distância de exclusão). A intensidade da repulsão entre duas moléculas de água
5.2. HAMILTONIANA
Capítulo 6
Resultados e discussões
As simulações foram realizadas em uma caixa de paredes rígidas de dimensões 100d×100d
e 40d×40d. Como é comum nesse tipo de modelo, um Passo de Monte Carlo (PMC)
corres-ponde ao número de tentativas que seria necessário para mover uma única vez cada molécula do sistema. Logo, um PMC para um sistema com 100 moléculas de água e 10 anfifílicos e 1 nanotubo corresponde a tentativa de se realizar 111 movimentos no sistema, não importando o tipo de movimento ou tentativa de movimento realizado.
6.1 Simulações das moléculas de água
Como já mencionado anteriormente, nesse modelo as moléculas de água são discos rígidos de
tamanho d. A distância máxima que uma molécula pode transladar em cada movimento foi
definido como 2d; logo, a distância média de deslocamento de uma “água”, de acordo com a
relação5.1.1éd. Cada molécula de água interage com as demais moléculas de água através de um potencial da forma da Figura5.7. Esse potencial possui um poço atrativo de energia −E0,
e uma parte repulsiva de energia 6E0. Assim, as águas adquirem, a baxas temperaturas, uma
configuração formando uma rede hexagonal. Essa configuração corresponde ao sólido formado pelas moléculas de água à temperaturas menores que a temperatura de fusão, o que está de acordo com a literatura [37].
Para o estudo da configuração de equilíbrio das moléculas de água, foram realizadas simula-ções de 400 águas em uma caixa 40d×40da temperaturaT =0,5. Este tamanho do sistema foi
escolhido devido ao fato de que em baixas temperaturas o tempo de relaxação é muito grande, despendendo muito tempo de simulação. Esse número de moléculas de água foi escolhido para que, de acordo com o potencial adotado, o comportamento das moléculas em altas temperaturas se assemelhasse ao de um líquido; menores concentrações nos dariam um sistema “rarefeito”, cujo comportamento assemelha-se ao comportamento de um gás.
6.1. SIMULAÇÕES DAS MOLÉCULAS DE ÁGUA
partir da transformada de Fourier:
F(ω) =
+∞
Z
−∞
exp(−iωr)g(r)dr (6.1)
Para a análise da relaxação da energia foram realizadas simulações de duas configurações inici-ais correspondentes a maior e a menor energia possível para esse sistema. O tempo de relaxação foi encontrado calculando o número de passos de MC onde ocorreu o encontro das energias das duas simulações. Isso ocorreu por volta de 4.106 passos. A Figura 6.1 mostra o gráfico da
energia em função do tempo de Monte Carlo.
Figura 6.1: Energia em função do tempo de MC de um sistema com 400 águas em uma caixa de tamanho 40 e temperaturaT =0,5. O destaque mostra a região onde ocorreu o encontro das curvas de energia das simulações
Figura 6.2: 400 águas em uma caixa quadrada 20×20 com T =0,5. À esquerda:
configu-ração inicial de maior energia. No centro: configuconfigu-ração inicial de menor energia. À direita: amostragem da configuração de equilíbrio (configuração “final”).
6.1. SIMULAÇÕES DAS MOLÉCULAS DE ÁGUA
Figura 6.3: Função distribuição radial do resultado da simulação de 400 águas comT =0,5.
T =0,5. A Figura 6.3 mostra o resultado da função distribuição radial para a configuração
de equilíbrio. A função distribuição radial foi calculada no intervalo de r=0 a r=L/2 com
incrementos∆r=0,1. A Figura6.4 mostra a transformada de Fourier deste resultado, obtida
utilizando o mesmo incremento. O pico na posição n=0,5 no gáfico daDFT mostra a
ocor-rência de peridiocidade. A posição do pico corresponde ar=2, concordando com o potencial
adotado.
Figura 6.4: Transformada de Fourier deg(r).
Simulações desse sistema foram realizadas em temperaturas no intervalo deT =0,5 aT =
1,4 em intervalos de temperatura∆T =0,1 no intuito de conhecer a temperatura de fusão da
água. O pico característico da Figura6.4 permanece em destaque até a temperatura de 1,0. A
partir desse valor a intensidade desse pico é de pouca relevância, indicando que a água funde
a temperatura por volta de T =1,0. Logo, as simulações de anfifílicos e nanotubos foram
6.2. SIMULAÇÕES DAS MOLÉCULAS ANFIFÍLICAS NA PRESENÇA DE ÁGUA
Para uma análise da convergência em temperaturas mais elevadas foram realizadas simula-ções do sistema anterior emT =1 eT =2. A Figura6.5mostra a curva da energia em função
do tempo de MC para duas configurações iniciais, de maior e de menor energia, nessas tem-peraturas. Na temperaturaT =1 a convergência da energia ocorreu antes de 3·104 PMC e na
temperaturaT =2 ocorreu por volta de 3·103PMC mostrando quão sensível é esa
convergên-cia com relação à temperatura. Ainda na Figura 6.5 há uma amostragem da configuração do
sistema no equiíbrio na temperatuaT =2, ilustrando a fase líquida do sistema.
Figura 6.5: Energia em função do tempo de MC em temperaturasT =1 (em cima, à esquerda)
eT =2 (em cima, à direita). A figura de baixo corresponde a uma amostragem do sistema no
equilíbrio em temperaturaT =2.
6.2 Simulações das moléculas anfifílicas na presença de água
Nas simulações das moléculas anfifílicas foi escolhido L=100d como tamanho do sistema.
Essa escolha nos permite colocar 2500 águas na caixa de acordo com as considerações feitas na seção anterior. Esse tamanho se mostrou suficiente para obter os resultados desse trabaho. Ao se adicionar anfifílicos no sistema, há um detalhe importante que deve ser analisado: em
modelos em redes o número de partículas em um sitema de tamanho L′ é fixo devido ao
6.2. SIMULAÇÕES DAS MOLÉCULAS ANFIFÍLICAS NA PRESENÇA DE ÁGUA
ocupamos n pontos da rede que definem o anfifílico. Uma forma simples para expressarmos
o que ocorre é dizendo que n águas são retiradas do sistema ao adicionarmos um anfifílico.
Apesar de cada anfifílico do nosso modelo ser definido por cinco pontos no espaço separados por d, que é a mesma distância de exclusão das moléculas de água (d), ao adicionarmos um anfifílico devemos retirar apenas três águas. Essa diferença é explicada quando analisamos a Figura 6.6. Como a distância de equilíbrio entre as moléculas de água é 2d, no espaço entre duas moléculas de água cabem três partículas do anfifílico. Isso possui influência significativa no resultado: caso essa consideração não seja feita, ocorre a formação de vazios entre duas moléculas de água, aumentando de forma artificial a mobilidade dos anfifílicos. Dessa forma, se a densidade de anfifílicos fosse superior a um certo valor, a quantidade de anfifílicos livres no sistema diminuiria, discordando com resultados já encontrados [23,38,39,40].
Figura 6.6: .
Como já mencionado anteriormente, as moléculas anfifílicas se movem por 4 movimentos distintos: translação, rotação, reptação e pivotamento. O deslocamento máximo de uma mo-lécula no movimento de translação é∆sur f =0,5d. O ângulo máximo de giro adotado para o
movimentação foi±π
3. Os demais movimentos dispensam essas definições.
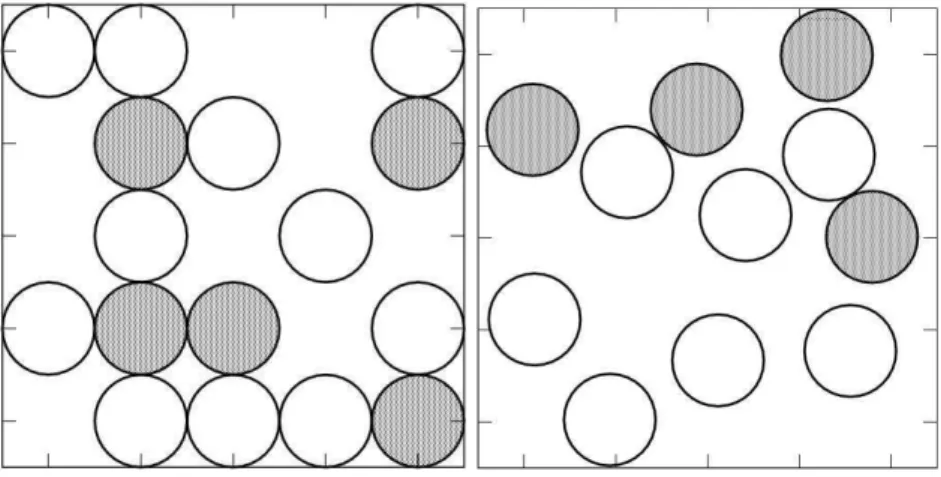
O potencial de interação entre as caudas de duas moléculas anfifílicas é ilustrado pela Figura 5.7B. A interação entre uma água e a cabeça de um anfifílico também é difinido por esse poten-cial. A interação entre a cauda de um anfifílico e a cabeça de outro é descrita pela Figura5.7C, que descreve, também, a interação entre a cauda de um anfifílico e uma água. Como resultado, ocorre a formação de estruturas de equilíbrio típicas observadas em soluções de anfifílicos. A Figura6.7ilustra a configuração inicial e uma configuração “final” (configuração típica do sis-tema no equilíbrio térmico) da simulação de 40 anfifílicos e 2380 águas na temperaturaT =3.
A Figura6.8mostra a ampliação da região em torno de uma micela deste mesmo resultado.
6.2. SIMULAÇÕES DAS MOLÉCULAS ANFIFÍLICAS NA PRESENÇA DE ÁGUA
Figura 6.7: Configuração inicial (acima) e final (abaixo) para um sistema contendo 40 anfifílicos
e 2380 águas na tempertatura T =3. Os círculos brancos representam as águas. Os círculos
pretos e cinzas representam, respectivamente, as cabeças e as caudas dos anfifílicos.
e (X1f ree) a concentração de anfifílicos livres definidos como [41]:
Xamph=
Namph
6.2. SIMULAÇÕES DAS MOLÉCULAS ANFIFÍLICAS NA PRESENÇA DE ÁGUA
Figura 6.8: Detalhe da Figura6.7.
Xi=
número de agregados comianfifílicos
Volume total(L×L) (6.3)
X1f ree=número de anfifílicos livres
1−3Xamph+3X1
(6.4)
Um anfifílico é dito pertencente a um agregado de anfifílicos se ele interage com algum an-fifílico deste agregado. Isso acontece se ao menos uma partícula de cada anan-fifílico se encontrar a uma distância menor que a distância de corte, definida como 3d em nosso trabalho.
A medida que se aumentaXamph,X1f reetambém aumenta linearmente para pequenas
concen-trações de anfifílicos. A partir de certa concentração a curva alcança um patamar de saturação e X1f ree permanece constante a medida que aumentamos oXamph. A Figura6.9 nos mostra o
resultado deX1f reeversusXamphpara diferentes temperaturas. Esse resultado foi obtido fazendo
uma média entre 40 amostras do sitema no equilíbrio térmico. A “distância” entre as amostras é de 4000 PMC.
A Figura6.9nos dá uma ideia da região onde se localiza a concentração micelar crítica. Na temperaturaT=2,0 esse valor é inferior a concentração 0,002, que corresponde a 20 anfifílicos nas simulações. Na temperaturaT =3,0 a cmc se encontra em torno da concentração 0,002.
Essa concentração corresponde a região de transição próxima a concentração micelar crítica. De fato, os resultados das simulações nessa concentração na temperaturaT =3,0 já mostram
formação de micelas.
Além de micelas, outras estruturas de equilíbrio podem ser obtidas neste modelo. A medida
que aumentamosXampho número médio de anfifílicos nos agregados também aumenta. Assim,
![Figura 3.1: Estrutura do grafite. Figura extraída da referência [8].](https://thumb-eu.123doks.com/thumbv2/123dok_br/15702714.629365/18.892.298.595.875.1054/figura-estrutura-do-grafite-figura-extraída-da-referência.webp)
![Figura 3.2: Célula unitária do diamante. Figura extraída da referência [9].](https://thumb-eu.123doks.com/thumbv2/123dok_br/15702714.629365/19.892.336.558.618.828/figura-célula-unitária-do-diamante-figura-extraída-referência.webp)

![Figura 3.8: Representação de uma micela. Figura extraída da ref [23]](https://thumb-eu.123doks.com/thumbv2/123dok_br/15702714.629365/27.892.242.678.124.335/figura-representação-de-uma-micela-figura-extraída-ref.webp)