J. Braz. Chem. Soc. vol.21 número10
Texto
Imagem
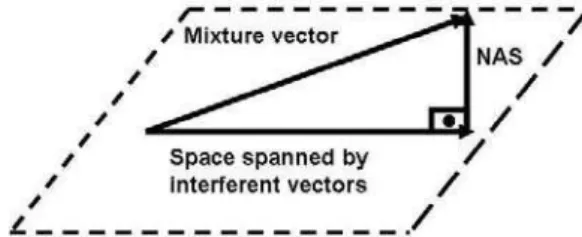
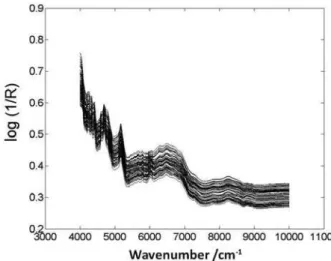
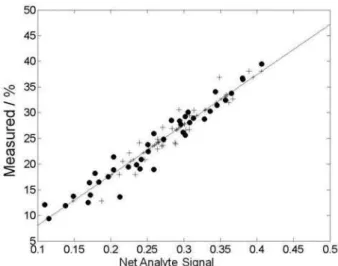
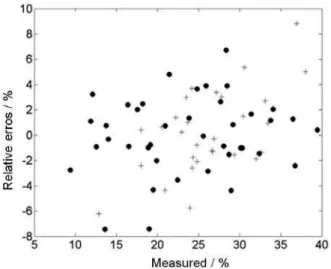
Documentos relacionados
In this sense, after steps 4 to 11 (Table 1) had been carried out, the microcomputer performed the titration ine adjustment by varying the volumes of the
Liposome Encapsulation of Lipophilic N -Alkyl-propanediamine Platinum Complexes: Impact on their Cytotoxic Activity and Inluence of the Carbon Chain Length.. Heveline Silva, a
The capability of the hybrid polymer to form inclusion complexes was evaluated by complexation with phenolphthalein (PP), which was determined by the decrease of
In short, the speciic reversible capacity of electrodes prepared with carbonaceous materials resulting from SB determined under galvanostatic conditions is
Light emission peaks obtained in the chemiluminescence reaction of ozone with free NO, released in the reduction of nitrogen- containing products of oxidized linoleic acid
Sterically-hindered fenchyl-substituted aromatic alkene 1a can be prepared using the Barton-Kellogg method from thiofenchone and a diazoanisole; whereas enol-ethers 1b and 1c
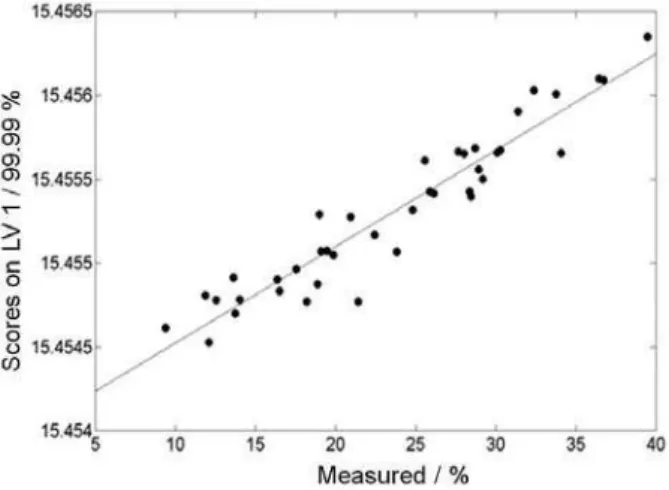
The data were subjected to principal component analysis to verify eventual similarities among the samples as well as to determine correlations between the elements and
The internal plasticization of chitosan with covalently linked long aliphatic branches, typically 12C, was accomplished through the condensation of the amino groups of chitosan
