Revista
Brasileira
de
Hematologia
e
Hemoterapia
Brazilian
Journal
of
Hematology
and
Hemotherapy
w w w . r b h h . o r g
Original
article
Study
of
enzyme
replacement
therapy
for
Gaucher
Disease:
comparative
analysis
of
clinical
and
laboratory
parameters
at
diagnosis
and
after
two,
five
and
ten
years
of
treatment
Ana
Maria
Almeida
Souza
∗,
Thiago
Pimentel
Muniz,
Rafael
Maciel
Brito
UniversidadeFederaldoPará(UFPA),Belém,PA,Brazil
a
r
t
i
c
l
e
i
n
f
o
Articlehistory:
Received7September2013
Accepted17March2014
Availableonline28May2014
Keywords:
Gaucherdisease
Enzymereplacementtherapy
Anemia Splenomegaly
a
b
s
t
r
a
c
t
Objective:ToevaluatetheimpactofenzymereplacementtherapyforGaucherDiseaseon
clinicalandlaboratoryparametersaftertwo,fiveandtenyearsoftreatment.
Methods:DatawerecollectedfrompatientrecordsandanalyzedusingBioEstatsoftware
(ver-sion5.0).Student’st-test,AnalysisofVariance(ANOVA),WilcoxontestandKruskal–Wallis
testwereusedforstatisticalanalysis.Hepatomegalyandsplenomegalywereanalyzedusing
theKappatest.
Results:Therewasasignificantincreaseinhemoglobinlevels(p-value<0.01)andplatelet
counts(p-value=0.01)withintwoyearsoftherapy.Atthesametime,thefrequenciesof
splenomegaly(p-value<0.01)andhepatomegaly(p-value<0.05)reduced.Theseresultswere
similaratfiveandtenyearsofenzymereplacementtherapy.
Conclusions: There are substantial and quick (withintwo years)laboratory and clinical
responsestoenzymereplacementtherapy.Theseimprovementscontinueaslongasenzyme
replacementtherapyisadministeredeverytwoweeks,asrecommendedbytheliterature.
©2014Associac¸ãoBrasileiradeHematologia,HemoterapiaeTerapiaCelular.Published
byElsevierEditoraLtda.Allrightsreserved.
Introduction
GaucherDisease(GD)isarecessiveautosomalhereditary
dis-orderclassifiedasaninbornerrorofthemetabolism.Itisthe
commonestlysosomalstoragediseaseandwasthefirstone
forwhichaspecifictreatmentwasdeveloped.Itoccursdueto
∗ Correspondingauthorat:InstitutodeCiênciasdaSaúde,FaculdadedeMedicinadaUniversidadeFederaldoPará,Prac¸aCamiloSalgado,
1,Umarizal,66050-060Belém,PA,Brazil.
E-mailaddress:[email protected](A.M.A.Souza).
adeficiencyintheactivityoftheenzyme-glucosidaseandis
characterizedbytheintra-lysosomalaccumulationof
gluco-cerebrosideinreticuloendothelialsystemcells.1Theenzyme
deficiencyiscausedbyamutationinthe-glucosidasegene,
locatedon chromosome1(GBA1).2 GDisararepan-ethnic
disorder,butitpresentsahighincidenceamongAshkenazi
Jews.Theworldwideincidenceisestimatedatfrom1:50,000
http://dx.doi.org/10.1016/j.bjhh.2014.05.005
1516-8484/©2014Associac¸ãoBrasileiradeHematologia,HemoterapiaeTerapiaCelular.PublishedbyElsevierEditoraLtda.Allrights
to1:100,000livenewborns,3whereastheincidenceinthe
Jew-ishpopulationrangesfrom1:400to1000livenewbornsinthe
USA.4
Clinically,GDpresentsawidevarietyofsignsand
symp-toms and is classified as non-neuronopathic (Type 1) or
neuronopathic (Types 2 and 3). Type 1 GD (95% of cases)
usually manifests with splenomegaly, hepatomegaly,
ane-mia,thrombocytopenia,bonedisease and delayedgrowth.5
Type2ischaracterizedbyaprecocious andfast brainstem
degeneration;3 thesepatients donot respondtotreatment
and death occurs within the first two years of life.6 Type
3GDpatientshaveaslow evolvingneurologicdisease and
usuallypresentwithseizures,eyemovementabnormalities
andmildsystemicinvolvementwithmeansurvivalbeingto
the thirddecade oflife.7 ThetreatmentofGD isbasedon
enzymereplacementtherapy(ERT),initiallybyalglucerase,8
butthiswaswidelysubstitutedbyitstherapeuticequivalent,
imiglucerase.9 Accordingly to Brazilian Ministry of Health,
therewere610ERT-dependentpatientsinthecountryin2010.
Therearenodataconcerningthenationalincidenceofthe
disease.10
Theobjectiveofthisstudywastoevaluatetheimpactof
ERTonclinical andlaboratory parametersofGDthrough a
comparativeanalysisofdataatdiagnosisandaftertwo,five
andtenyearsoftreatmentinapopulationfromParáState,
Brazil.
Methods
Thiswasananalyticalobservationallongitudinal
retrospec-tive study (historical cohort) of patients at the Fundac¸ão
Centro de Hemoterapia e Hematologia do Pará (HEMOPA),
Belém, Pará State. Thepatients were diagnosed with
non-neuronopathic(Type1) andneuronopathic (Type3)GDand
weretreatedandfollowed-upatHEMOPAbetween2000and
2011.
Theinclusioncriteriaforthe studywere tohavea
con-firmeddiagnosisofGDandtobetreatedandfollowed-upat
HEMOPAforatleast24months.Clinicalrecordspriorto
treat-mentwerealsonecessary.
Datawerecollectedfrompatientrecordsusinga
question-nairedesignedforthisstudy.Thisquestionnaireincludedthe
followingitems:demographiccharacteristics,geneticprofile,
ERT dose, hematological aspects (hemoglobin levels, white
cell count and platelet count) and clinical manifestations
(splenomegaly, hepatomegaly and neurologicalsymptoms).
Datawerecollectedatfourdifferenttimepoints:atdiagnosis
andaftertwo,fiveandtenyearsofERT.Diagnosiswasdefined
asthe time whenGaucher’s cells and/orthe -glucosidase
deficiency were identified. Data concerning the two-,
five-and ten-year timepoints were collected based onthe first
administrationofERT.ThisstudywasapprovedbytheEthics
CommitteeofHEMOPA(register#0016.0.324.324-11).
Statisticalanalysis
Data were placed in tables and graphs drawn using the
MicrosoftExcel2010software.ERTdoses,hemoglobinlevels,
whitecellcountandplateletcountovertimewereanalyzed
usingStudent’st-test,AnalysisofVariance(ANOVA),Wilcoxon
and Kruskal–Wallis tests, as applicable. Splenomegaly and
hepatomegalywereanalyzedusingtheKappatest.Allresults
withp-values<0.05wereconsideredsignificantandtestswere
carriedoutusingtheBioEstat(version5.0)software.
Results
Demographiccharacteristics
Recordsof24patientsdiagnosedwithGDwerefoundandof
these,13mettheinclusioncriteria.Eightwerefemale(61.50%)
andfiveweremalepatients(38.50%).Agesrangedfromfourto
43years(mean:24.53years).Thirteenpatients(100%)wereon
ERTfortwoyears;nine(69.23%)hadcompletedfiveyearsof
treatmentandsixpatients(46.15%)hadbeentreatedforten
years.Inordertopreservepatients’identity,theyare
num-bered1through13.
Atdiagnosis,themeanageofpatientswas13.49yearsold
(±30.10 years). Eightpatients (61.53%) wereunder 12 years
old atdiagnosis(patients1, 3,4,5,8, 11,12 and 13),three
(23.07%)werewomenover12years(patients6,9and10)and
two(15.4%)weremenover12years(patients2and7).Only
onepatient(7.7%)wasdiagnosedwithType3GD;thispatient
diedatage22aftertenyearsoftreatment.Allotherpatients
werediagnosedasType1.
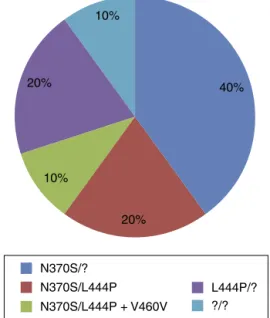
Geneticprofile
Tenpatients(76.92%)weresubmittedtoGBA1mutation
anal-ysiswiththefrequenciesofthemutationslistedinFig.1.
ERTdose
ERT wasadministered every twoweeks.Table1shows the
amount ofenzyme(IU/kg)giventoeachpatientduringthe
studyintervals.
N370S/?
40%
20% 10%
10% 20%
N370S/L444P N370S/L444P + V460V
L444P/? ?/?
Table1–Enzymereplacementtherapydose
administered(IU/kg)topatientswithGaucherDisease bystudyinterval.
Patient Atstart 2years 5years 10years
1 27.9 25.0 28.0 30.0
2 30.0 30.0 26.0 –
3 36.6 40.0 – –
4 30.0 30.0 – –
5 18.4 42.1 – –
6 33.3 37.1 30.0 15.0
7 44.0 44.0 41.0 37.0
8 22.0 61.0 54.0 –
9 35.0 24.6 27.0 15.0
10 15.0 29.0 29.0 –
11 37.7 37.7 26.9 15.0
12 32.4 40.0 – –
13 29.4 13.6 40.0 85.7
Therewasnostatisticaldifferenceinthedosereceivedbetween anyofthestudyintervals(p-value=0.78).Meandoseofallperiods togetherwas32.88IU/kg.
Laboratoryparameters
Table2 presents hemoglobinlevels (g/dL) atdiagnosis and
aftertwo,fiveandtenyearsoftreatment.
Themeanhemoglobinlevelatdiagnosis(9.9g/dL)andafter
twoyearsoftreatment(12.4g/dL)werecomparedusing
Stu-dent’st-test.Themeandifferencebetweenthesetwoperiods
(2.5g/dL) was statistically significant (p-value=0.0039; 95%
confidenceinterval: 0.0762–4.15). ANOVA, however, did not
showstatisticaldifferencewhentheintervalsof2,5and10
yearswerecompared.Ontheotherhand,hemoglobinlevels
at5and10yearsweresignificantlyhigherthanthelevelsat
diagnosis(p-value<0.01forbothperiods).
Table3showsthe totalwhitecell countinthedifferent
studyintervals.
No statistical significance was found for the white cell
countbetweenanyofthestudyintervals.
Table4presentstheplateletcountsatdiagnosisandafter
two,fiveandtenyearsofERT.
Table2–Hemoglobinlevels(g/dL)ofpatientsunder enzymereplacementtherapyforGaucherDiseaseby studyinterval.
Patient Atdiagnosis 2years 5years 10years
1 10.0 14.0 11.4 13.4
2 8.2 16.1 13.9 –
3 10.5 9.6 – –
4 12.2 11.8 – –
5 12.6 13.8 – –
6 10.6 12.0 10.5 12.2
7 12.8 12.2 13.4 13.9
8 9.5 11.9 11.3 –
9 8.2 13.6 13.8 14.0
10 9.4 10.0 12.3 –
11 7.4 13.8 13.6 14.6
12 8.7 10.7 – –
13 9.0 11.4 12.5 10.5
Table3–Whitecellcount(×109/L)ofpatientsunder enzymereplacementtherapyforGaucherDiseaseby studyinterval.
Patients Atdiagnosis 2years 5years 10years
1 4.20 3.40 3.13 4.35
2 10.60 9.93 9.58 –
3 1.80 1.06 – –
4 4.56 6.30 – –
5 6.71 5.66 – –
6 11.30 11.70 13.80 13.10
7 5.50 15.40 10.80 10.70
8 12.30 17.80 6.91 –
9 4.40 5.30 4.81 7.00
10 8.80 5.80 6.50 –
11 3.00 2.90 3.88 4.23
12 6.18 9.30 – –
13 2.90 3.00 3.94 3.00
Thedifferenceinplateletcountsbetweendiagnosis and
aftertwoyearsoftreatment,evaluatedusingthe Wilcoxon test(non-normaldistributionatdiagnosis),wasstatistically significant(p-value=0.01).Thedifferencesbetweentheother periodswerenotstatisticallysignificant.
Clinicalmanifestations
On analyzing splenomegaly, two patients were excluded
becausetheyhadbeensubmittedtosplenectomypriortoERT. Allofthe11remainingpatientspresentedsplenomegalyat diagnosis, detectedeitherbyphysical exam or radiological imaging.AftertwoyearsofERT,63.63%ofthecasespresented withsplenomegaly.Thisreductionwasstatisticallysignificant (p-value<0.05).Areduction,albeitnon-significant,wasfound
inthe frequencyofsplenomegalycomparing patientsafter
onlytwoyearsoftreatmentandafterfiveandtenyearsof treatment.
Similar resultswerefoundconcerninghepatomegaly.Of
the 13 patients, 69.23% had hepatomegaly atdiagnosis.At
theendoftwoyearsofERT,only30.77%continuedwithany degree ofhepatomegaly. Thisreduction wassignificant (p -value<0.05).Therewasafurtherreductioninthenumberof
Table4–Plateletcount(×109/L)ofpatientsunder enzymereplacementtherapyforGaucherDiseaseby studyinterval.
Patients Atdiagnosis 2years 5years 10years
1 90.0 87.0 74.9 121.0
2 105.0 348.0 378.0 –
3 20.0 20.3 – –
4 240.0 205.0 – –
5 117.0 165.0 – –
6 390.0 336.0 317.0 127.0
7 200.0 232.0 222.0 312.0
8 112.0 258.0 204.0 –
9 87.0 165.0 178.0 186.0
10 120.0 154.0 128.0 –
11 59.0 101.0 71.4 131.0
12 92.4 158.0 – –
patientswithhepatomegalyat5and10yearsoftreatment,
butthedifferenceswerenotstatisticallysignificant.
Neurologicalsymptomswere noticedonlyintheType 3
patientinthestudysample.Thispatientpresentedseizures,
tremorsoftheextremities,dyslalia anddifficultieswalking
duringalltheperiodofERT(10years).
Discussion
GDisusuallydescribedasapan-ethnicdisease,attackingmen
and womeninequalproportions. However,this study
pre-sentedahigherprevalenceoffemale(61.5%)comparedtomale
patients(38.5%).ThisdataissimilartoastudybySobreira&
Bruniera,3whoshowedahigherprevalence(67.8%)offemale
patients,anddiffersfromFerreiraetal.,1whoreportedaslight
predominanceofmalepatients.Thisisattributedtothesmall
samplesizecommonwhenstudyingrareconditions.Nodata
relatingthepatientstoAshkenaziparentagewerefound.
Mean age at diagnosis was slightly higher than that
reportedbySobreiraandBruniera(11.8yearsold)3andlower
than that inthe study ofCharrow et al. (17.4years old).11
Giraldoetal.12reportedameanageatdiagnosisof27.4years
old.Earliermanifestationsofthediseasearetypicalfor
Latin-Americanpopulations;inthesepatients,thefirstsymptomsof
GDoccurat11yearsofageandcanbemoresevere.13Such
dif-ferencesshowthatGDcanpresentatdifferingagesindifferent
populationsandisanexpressionofthedisease’s
heterogene-ity.
TheN370S mutation,in 70% ofthe individuals
submit-tedtoGBA1 analysis, wasthe mostcommon inthis study
sample.Thisdataissimilartoastudyof1698patientsfrom
theGaucher Registry,11 inwhich 84% ofindividuals had at
leastoneN370Sallele;however,23% were homozygousfor
this mutation, different tothe current study, in which no
homozygousindividuals werefound. Thesecond most
fre-quent mutation was L444P, which occurred in 50% of the
sample.Thisfrequencyisslightlyhigherthanthatof
Char-rowetal.(30%),11Kaplanetal.(34%)5andconsiderablyhigher
thanthatofAnderssonetal.(23.7%).14Evenso,no
homozy-gousindividualswiththeL444Pmutationwerefoundinthis
sample.Noothermutationthatisconsideredcommoninthe
literature,6suchas84GGandIVS+1,wasfound.Ontheother
hand,theV460Vmutation,whichisnotconsideredacommon
mutation,wasassociatedtotheL444Pmutation.
Thedivergencesbetweenthesedataaremostlikelydueto
thefactthattheaforementionedstudiesgathered
informa-tionfromfivecontinentsandthatLatin-Americarepresented
asmall portionofthetotal.AstudyofjustLatin-American
patient records inthe Gaucher Registry13 founda low
fre-quency of L444P homozygosis. In the same study, 82% of
individuals hadatleast oneN370Sallele,but onlyasmall
portion ofthem was homozygous. A small study in Santa
Catarina State, Brazil1 corroborates these findings; 60% of
individualswere heterozygousfortheN370Smutationand
therewas onlyone homozygouscase.TheL444P mutation
was found in 30% of individuals and there was one case
ofhomozygosis. Thedifferencesbetween studies inBrazil,
Latin-AmericaandworldwideshowthatBrazilianand
Latin-American patients have a genetic profilethat differs from
that of patients from other nations with such differences
beingexpressedasdiversephenotypic presentationsofthe
disease.
Furthermore,astudywith48Brazilianpatientsfrom
dif-ferentregionsofthecountryfoundsevendifferentpreviously
unknown mutated sequences of GBA. Five were missense
changes(S125N,F213L,P245T,W378C,D399H),onewasan
in-frameinsertionandtheotheronewasasplicingmutationin
acomplexallele(L461P+IVS10+1G>T).Thesedatareinforce
thegeneticheterogeneityofBrazilianGDpatientsaswasalso
foundinthecurrentresearch.Thisstudyalsosuggeststhat
thepresenceoftheN370Sallelewouldbeaprotectivefactor
forneurologicalmanifestations.15Indeed,theonlyType3GD
patient presentedthe L444Pallele,alongwithanunknown
allele.
TheERTinfusionperiodicitywasconstantduringall
inter-vals of the treatment. The mean dose administered was
slightly higherthan the dose recommended by the
Brazil-ian Consensusfor the treatment ofGD (32.88IU/kg versus
30.0IU/kg).7Thissmalldifferenceisduetothefactthatthe
doseregisteredinpatients’recordswasthetotalamountand
thedivisioninrespecttothepatients’weightfrequentlydid
notresultinanexact value.Furthermore,individual
varia-tionsobservedinTable1reflect:1–thelevelsoftheseverity
ofthediseasethatrequirehigherorlowerdoses,asexplained
by Goker-Alpan;16 2– theneed ofnewadjustmentsto the
doseduetoadramaticreductioninthestockofimiglucerase
afteraviralinfectionthatoccurredin2009;173–theretake
ofthedosesrecommendedbytheTherapeuticGuidelinesfor
GDoftheBrazilianMinistryofHealth,18afterthestockwas
replenished.
In the study sample, ERT was effective in raising the
hemoglobinlevelsinthefirsttwoyearsoftreatmentandin
keepingthemstabileduringalltheperiodthatthe enzyme
wasadministered.Thiswasalsotrueevenwhenthepatients
wereunder12yearsoldatdiagnosis.Theimprovementin
ane-miaandthestabilizationofhemoglobinlevelshavealready
been demonstrated in a two-yearstudy of148 patients in
whichthenumberofanemicpatientsdrasticallyfellwithin
the first six months of starting ERT.3 The results of the
hemoglobin levels at the end of two years of treatment
are in accordance with the therapeutic goal proposed by
Weinrebetal.,19i.e.,hemoglobin≥11g/dLinunder
12-year-old patients (87.5%in thecurrent study),≥11g/dL inadult
women(notachievedbyonlyonewomaninthisstudy)and
≥12g/dLinadultmen(achievedbybothmeninthecurrent
study).
Theplateletcountalsorosesignificantlybytheendoftwo
years of ERT. Weinrebet al.19 divided the therapeutic goal
forplatelet countintothreegroups: patientswithcountat
diagnosis>120.0×109/Lmustremaininthisrange,patients
withcountsrangingfrom 60.0×109/Lto120.0×109/L must
achieve countsover120.0×109/L, andpatientswithcounts
<60.0×109/L must increase their count by twofold. In the
study sample,the results were satisfactory, asnine of the
13patients(69.23%)achievedthetherapeuticgoalwithintwo
yearsoftreatment.Forthefirsttwogroups,onlyPatient1,who
belongedtothesecondgroup,didnotreachthetherapeutic
goal.However,noneofthepatientsinthethirdgrouptripled
presentamoreseverediseaseandrespondslowlyand
gradu-ally(orevenunsatisfactorily)totherapyasfarastheplatelet
countisconcerned.5
Nonetheless,ERTiscapableofkeepingthediseasestable
overtheyearsinmostpatients.Thisisevidencedbythe
nor-malmeanhemoglobinlevelsandplateletcountsattheendof
fiveandtenyearsoftreatment.Thiseffectwasobservedina
studywith887childrenwhosehemoglobinlevelsandplatelet
countsimprovedinthefirsttwoyearsandremainedstable
overeightyearsofobservation.14Despitethepresenceofadult
patientsinthestudysample,ourresultssuggestthatthe
effi-cacyofERT issimilar indifferentagegroupsinrelationto
thesetwohematologicalparameters.
Additionally,theresultssuggestthatalower-doseregimen
(i.e.,approximately30IU/kgeverytwoweeks)isaseffectivein
reachingthetherapeuticgoalsasahigh-doseregimen,as
orig-inallyproposedbyBartonetal.8 (60IU/kgeverytwoweeks).
Thisbringsaneconomicadvantageinnon-wealthynations,
suchasBrazil,allowingthemtoreservehigh-doseregimens
forcriticallysymptomaticpatientswhoneedafasterrecovery,
similartowhatisreportedinIsrael.20
Theabsence ofstatistical significance inthe white cell
countisprobablyduetoextremelyvariableresultsbetween
individuals, i.e., while some presented leukopenia,
oth-erspresentedleukocytosis.Leukocytosis is notunexpected
in GD patients as they are more likely to acquire
infec-tions. Furthermore, the leukocyte function is disturbed in
GDduetosubstrate accumulationinthesecells,especially
the macrophage/monocyte lineage, inherenttothe disease
physiopathology.21 Additionally, leukopenia is usually not
severe and rarely requires any intervention, making the
whitecellcountanon-specificparameterinthefollow-upof
patients.6
SimilartothestudybyFerreiraetal.,1splenomegalywas
foundin100%ofpatients(splenectomizedpatientswerenot
included)whenclinical and radiological methodstodetect
thisalterationareconsidered.Despitethesignificant
reduc-tioninthenumberofpatientspresentingwithsplenomegaly
after two years of ERT, it was not possible to determine
whethertherewasareductioninvolumeinpatientswhostill
presentedthis alteration,asmostrecordsdidnotstatethe
degreeofsplenomegalyand,whentheydid,thedatawerenot
standardized,precludingamoreaccurateanalysis.
Consid-eringthatinsubsequentperiodstherewasreductioninthe
numberofpatientswithsplenomegaly,eventhoughnot
statis-ticallysignificant,theauthorsbelieveareductioninthespleen
volumealsooccurredinthefirsttwoyearsofERT,similarto
whatisreportedintheliterature.
Thenumberofpatientswithhepatomegalyhaddecreased
bytheendoftwoyearsofERTsimilartothestudyofSobreira
&Bruniera;3however,inthatstudy,thereductioninliver
vol-umeoccurredprimarilybetweenthe12thand18thmonths
ofERT.Therewasnostatisticallysignificantreductioninthe
followingperiodswhichshowstheearlyresponsetoERTin
respecttohepatomegaly(within2years).
ERTwasinefficienttocontrolneurologicalsymptoms in
theonlyType3patientinthestudy.Symptomsofthispatient
persisteddespiteincreasesindose.Thiscaseexemplifiesthe
higherseverityofType 3GDasalreadyreported by
Goker-Alpan16andCox.17
Conclusion
ERThasanimportantimpactonimprovinghemoglobinlevels
andplateletcounts.Theseparametersrespondtotreatment
inthe initialphase ofdrug treatment(within 2years) and
the responseismaintainedaslong asERT isadministered
every two weeksaccordinglyto the standardregimen
pro-posedintheliterature.Hepatomegalyandsplenomegalyalso
respondsignificantlytoERTwithintwoyearsoftreatment.As
splenomegalyisthemostfrequentmanifestation,the
pres-enceofthissigninapatientwithcytopeniashouldleadthe
attendingphysicianto considerGD asoneofthe
differen-tialdiagnosis.Due tothewiderangeofmanifestations,the
treatmentofGDandfollow-upmustbeperformedbya
mul-tidisciplinaryteam.
Conflicts
of
interest
Theauthorsdeclarenoconflictsofinterest.
r
e
f
e
r
e
n
c
e
s
1.FerreiraJS,FerreiraVL,FerreiraDC.EstudodaDoenc¸ade GaucheremSantaCatarina.RevBrasHematolHemoter. 2008;30(1):5–11.
2.JmoudiakM,FutermanAH.GaucherDisease:pathological mechanismsandmodernmanagement.BrJHaematol. 2005;129(2):178–88.
3.SobreiraEA,BrunieraP.Avaliac¸ãodedoisanosdetratamento dadoenc¸adeGauchertipo1comterapiadereposic¸ão enzimáticaempacientesdoestadodeSãoPaulo.BrasilRev BrasHematolHemoter.2008;30(3):193–201.
4.OliveiraFB[dissertation]Avaliac¸ãodaQualidadedeVidade PacientescomDoenc¸adeGaucher.Doenc¸adeFabrye Mucopolissacaridoses.PortoAlegre(RS):UFRGS;2010.
5.KaplanP,AnderssonHC,KacenaKA,YeeJD.Theclinicaland demographiccharacteristicsofnonneuronopathicGaucher Diseasein887childrenatdiagnosis.ArchPediatrAdolesc Med.2006;160(6):603–8.
6.PastoresGM,HughesDA.GaucherDisease.GeneRev.2011
http://www.ncbi.nlm.nih.gov/books/NBK1269/
7.MartinsAM,LoboCL,SobreiraEA,ValadaresER,PortaG, SemionatoFilhoJ,etal.Tratamentodadoenc¸adeGaucher: umconsensobrasileiro.RevBrasHematolHemoter. 2003;25(2):89–95.
8.BartonNW,BradyRO,DambrosiaJM,BisceglieAM,Doppelt SH,HillSC,etal.Replacementtherapyforinheritedenzyme deficiency–macrophagetargetedglucorebrosidasefor Gaucher’sDisease.NEnglJMed.1991;324(21):1464–70.
9.GrabowskiGA,BartonNW,PastoresG,DambrosiaJM, BanerjeeTK,McKeeMA,etal.EnzymetherapyinType1 GaucherDisease:comparativeefficacyof
mannose-terminatedglucocerebrosidasefromnaturaland recombinatsources.AnnInternMed.1995;122(1):33–9.
10.SouzaMV,KrugBC,PiconPD,SchwartzIV.Medicamentosde altocustoparadoenc¸asrarasnoBrasil:oexemplodas doenc¸aslisossômicas.CienSaudeColet.2010;15(Suppl3): 3443–54.
12.GiraldoP,AlfonsoP,AtutxaK,Fernández-GalánMA,BarezA, FrancoR,etal.Real-worldclinicalexperiencewithlong-term miglustatmaintenancetherapyintype1Gaucherdisease: theZAGALproject.Haematologica.2009;94(12):1771–5.
13.DrelichmanG,LinaresA,VillalobosJ,CabelloJF,Kerstenetzky M,KohanRM,etal.EnfermedaddeGaucherEn
Latinoamérica–UnInformeDelRegistroInternacionalYDel GrupoLatinoamericanoParaLaEnfermedaddeGaucher. Medicina(BAires).2012;72(4):273–82.
14.AnderssonH,KaplanP,KacenaK,YeeJ.Eight-yearclinical outcomesoflong-termenzymereplacementtherapyfor884 childrenwithGaucherDiseaseType1.Pediatrics.
2008;122(6):1182–90.
15.SiebertM,BockH,Michelin-TirelliK,CoelhoJC,GiuglianiR, Saraiva-PereiraML.Novelmutationsinthe
glucocerebrosidasegeneofBrazilianpatientswithGaucher disease.JIMDRep.2012;9:7–16.
16.Goker-AlpanO.OptimaltherapyinGaucherdisease.Ther ClinRiskManag.2010;6:315–23.
17.CoxTM.GaucherDisease:clinicalprofileandtherapeutic developments.Biologics.2010;4:299–313.
18.Brazil.GaucherDisease.MinistryofHealth.Secretaryof AttentiontoHealth,OrdinanceSAS/MSn◦708Clinical
ProtocolandTherapeuticGuidelines;2011.
19.WeinrebN,TaylorJ,CoxT,YeeJ,vonDahlS.Abenchmark analysisoftheachievementoftherapeuticgoalsfortype1 Gaucherdiseasepatientstreatedwithimiglucerase.AmJ Hematol.2008;83(12):890–5.
20.TukanI,Hadas-HalpernI,AltarescuG,AbrahamovA,Elstein D,ZimranA.Achievementoftherapeuticgoalswithlow-dose imigluceraseinGaucherDisease:asinglecenterexperience. AdvHematol.2013:151506.Epub.