w w w . r b h h . o r g
Revista
Brasileira
de
Hematologia
e
Hemoterapia
Brazilian
Journal
of
Hematology
and
Hemotherapy
Original
article
Clinical,
hematological
and
genetic
data
of
a
cohort
of
children
with
hemoglobin
SD
Paulo
do
Val
Rezende
a,∗,
Kenia
da
Silva
Costa
a,
Jose
Carlos
Domingues
Junior
a,
Paula
Barezani
Silveira
a,
André
Rolim
Belisário
a,
Celia
Maria
Silva
a,
Marcos
Borato
Viana
baFundac¸ãoHemominas,BeloHorizonte,MG,Brazil
bUniversidadeFederaldeMinasGerais(UFMG),BeloHorizonte,MG,Brazil
a
r
t
i
c
l
e
i
n
f
o
Articlehistory:
Received9April2016
Accepted2May2016
Availableonline21May2016
Keywords:
Sicklecelldisease
HemoglobinS/D-Punjab
HemoglobinS-KorleBu
Children Haplotypes
a
b
s
t
r
a
c
t
Introduction:ThehemoglobinFSDisveryuncommoninnewbornscreeningprogramsfor
sicklecelldisease.IntheprogramofMinasGerais,Brazil,theclinicalcourseofchildren
withhemoglobinSDwasobservedtobeheterogeneous.Theobjectiveofthisstudywas
toestimatetheincidence(1999–2012)andtodescribethenaturalhistoryofacohortof
newbornswithhemoglobinSD.
Methods:Isoelectricfocusingwastheprimarymethodusedinnewbornscreening.
Poly-merasechainreaction-restrictionfragmentlength polymorphismandgene sequencing
wereusedtoidentifymutantallelesandforhaplotyping.Gap-polymerasechainreaction
wasusedtodetectalpha-thalassemia.
Results:ElevencasesofhemoglobinS/D-PunjabandeightofHbS-KorleBuweredetected.
OthervariantswithhemoglobinDmobilitywerenotidentified.AllhemoglobinD-Punjab
andhemoglobinKorleBualleleswereassociatedwithhaplotypeI.Amongthechildrenwith
hemoglobinS/D-Punjab,therewerefourwiththeSCARhaplotype,sixwiththeBenin
haplotype,andoneatypical.ResultsoflaboratorytestsforhemoglobinS/D-Punjaband
hemoglobinS-KorleBuwere:hemoglobin8.0and12.3g/dL(p-value<0.001),leukocytecount
13.9×109/Land10.5×109/L(p-value=0.003),reticulocytes7.5%and1.0%(p-value<0.001),
hemoglobinFconcentration16.1%and6.9%(p-value=0.001)andoxygensaturation91.9%
and97%(p-value=0.002),respectively.OnlyhemoglobinS/D-Punjabchildrenhadacutepain
crisesandneededbloodtransfusionsorhydroxyurea.ThosewiththeBeninShaplotypehad
highertotalhemoglobinandhemoglobinFconcentrationscomparedtotheCARhaplotype.
TranscranialDopplerwasnormalinallchildren.
Conclusion:TheclinicalcourseandbloodcellcountsofchildrenwithhemoglobinS/D-Punjab
wereverysimilartothoseofhemoglobinSSchildren.Incontrast,childrenwithhemoglobin
S-KorleBuhadclinicalcourseandbloodcellcountslikechildrenwiththesicklecelltrait.
©2016Associac¸ ˜aoBrasileiradeHematologia,HemoterapiaeTerapiaCelular.Published
byElsevierEditoraLtda.ThisisanopenaccessarticleundertheCCBY-NC-NDlicense
(http://creativecommons.org/licenses/by-nc-nd/4.0/).
∗ Correspondingauthorat:RuaFreiGonzaga,301,30315–170BeloHorizonte,MG,Brazil.
E-mailaddress:[email protected](P.d.V.Rezende).
http://dx.doi.org/10.1016/j.bjhh.2016.05.002
1516-8484/©2016Associac¸ ˜aoBrasileiradeHematologia,HemoterapiaeTerapiaCelular.PublishedbyElsevierEditoraLtda.Thisisan
Introduction
Sickle cell disease (SCD)is apublic healthproblem
world-wide. The hemoglobin (Hb) SD subtype seems to be very
rare. ItincludestheHb S/D-Punjabvariantthat apparently
isassociatedwithamoresevereclinical course,and other
HbS-non-Punjabvariants,withlimitedinformationregarding
laboratoryandclinicaldata.1–3
TheHbD-PunjabvariantisthemostcommonD-subtype
describedintheliteratureworldwide.Itwasfirstdescribedin
1951byItano.4Itresultsfromthereplacementoftheamino
acidglutamatewithglutamine atposition121ofthe
beta-globinchain[beta121(GH4)Glu>Gln;HBB:c.364G>C].
AnothervariantwithinthewindowofHbDusing
isoelec-tricfocalization(IEF),butnotusinghigh-performanceliquid
chromatography(HPLC),isHb KorleBu,whichresultsfrom
thereplacementoftheaminoacidaspartatewithasparagine
atposition72ofthebeta-globinchain[beta73(E17)Asp>Asn;
HBB:c.220G>A].ItoriginatesfromthewesternregionofAfrica,
andits dissemination toAmericais probablylinkedtothe
slavetradeinthe17thto19thcenturies.5
TheclinicalcourseofpatientswithHbSDdiseaseseems
tobeheterogeneous, depending onthe Hb D variant. This
suggeststhatthe sicklingprocessislikelytheresultofthe
interactionbetweentheintracellularHbSandHbDvariants.
Thisinteractionmaybestrengthenedorweakened,
depend-ingontheHbvariantco-inheritedwiththeSmutation.6–14
Children with Hb S/D-Punjab disease seem to present a
clinical course similar to those with homozygous Hb SS
disease.8,10,11,15,16
Thus,patientswithHbS/D-Punjabdiseaseshouldreceive
the same treatment protocol as those with Hb SS disease
becausetheymay alsoexperience potentiallyfatal
compli-cationsduringtheirlives.10,11,17 Theuse ofhydroxyurea in
childrenwithHbSDdiseasehasbeenrestrictedtoisolated
cases.11,18,19
Theobjectiveofthisstudywastoestimatetheincidence
andtodescribethenaturalhistoryofnewbornswiththeHb
SDpatternbyIEF,screenedaspartoftheNeonatalScreening
ProgramintheBrazilianstateofMinasGerais(PTN-MG).
Addi-tionally,beta-globinclusterhaplotypesfortheS,D-Punjaband
KorleBumutationsweredeterminedinordertotrytotracethe
originoftherespectivemutations.
Methods
This descriptive study is based on a retrospective cohort.
Archived medical records from the Fundac¸ão Hemominas
(GovernmentBloodCenter)andthePTN-MGdatabankwere
used.IEFofdriedbloodspotsamples(NeonatalHemoglobin
ResolveScreenKit,PerkinElmerLifeandAnalyticalSciences,
Finland)hasbeentheprimarymethodofnewbornscreening
atPTN-MG.Allele-specificpolymerasechainreaction(PCR)for
A,S,C,andD-Punjaballeleswasintroducedintheblood
bankprotocolasaconfirmatorytestin2010.
Thepopulationinitiallycomprised21childrenwithHbFSD
atbirth.HbFSDmeansthatthechildrenwerebornwiththree
Hbfractions:Hb F(themajorfractionatbirththatsteadily
Molecular Markers
600 bp
A
B
500 bp 400 bp
300 bp
200 bp
C C T T T A G TG/A A T G G C C T G
1 2 3 4 5
572 bp
300 bp
272 bp
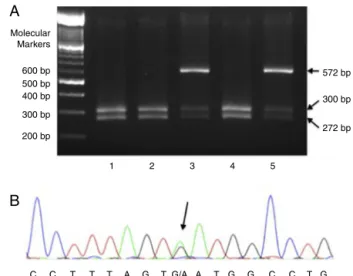
Figure1–MoleculardetectionofmutationsinHbD-Punjab
andHbKorleBu.(A)Polymerasechainreaction-restriction
fragmentlengthpolymorphismwithEcoRIinfivechildren
withHbD-Punjab.Patients1,2,and4hadwildalleles(two
bandsof300and272basepairs)andPatients3and5had
anadditionalbandof572basepairsthatindicatesa
heterozygousmutationinthecodon121ofHBB(Hb
D-Punjab);(B)electropherogramofachildwithHbKorleBu.
Thearrowpointstotheheterozygousmutation
HBB:c.220G>A(GAT>AAT;Asp>Asn)detectedthroughgene sequencing.
decreasesoverthefirstyearoflife),HbS,andanHbwitha
zoneDmobilityinelectrophoresis.Theywerebornbetween
January1,1999andDecember 31,2012;atotalof3,590,315
childrenwerescreenedinthis period.Thepatients’clinical
andlaboratorydatauptoDecember31,2014werereviewedso
thatallchildren,excepttwo,hadbeenfollowedupforatleast
twoyears.
Twopatientswereexcludedfromtheclinicalandmolecular
analyses because ethylenediaminetetraacetic acid
(EDTA)-anticoagulated blood samples had not been obtained for
moleculartests:onechildmovedabroadandanothermoved
outofthestate.Contactwiththefamilieshasfailedthusfar.
ThesechildrenwereusedonlytocalculatetheincidenceofHb
SDinthecohort.
Fragments of the beta-globin gene (HBB) containing
exon 3 (forward primer 5′-TCATGCCTCTTTGCACCATTC-3′;
reverseprimer5′-CACTGACCTCCCACATTCCC-3′)were
ampli-fied using PCR, and polymerase chain reaction-restriction
fragment length polymorphism (PCR-RFLP) was conducted
withtheEcoRIenzymetodetecttheD-Punjaballele(Figure1A).
Ifthereactionwasnegative,thethreeexonsoftheHBBgene
weresequencedtoidentifythemutationunderlyingtheother
Hbvariants(primercompositionavailableonrequest).DNA
sequencing was done in an ABI 3130 capillary sequencer
(Applied Biosystems, FosterCity, CA,USA). Figure 1B
illus-tratesthegenesequencingoftheregioninwhichtheHBB:
c.220G>AmutationunderlyingHbKorleBuislocated.
Detec-tionofsevenmorecommonHBAdeletionswascarriedoutby
The-globingeneclusterhaplotypingwascarriedoutby
PCR-RFLPofsix restriction sites: HindIIIin the IVS-IIof G␥
(rs113425530),HindIII inthe IVS-IIofA␥(rs28440105), HincII
in(rs10128556), HincII3′ to(without rs),HinfI 5′ to
(rs16911905),andHinfI3′to(rs10837631).Additionally,HincII
5′to
(rs3834466)andAvaIIatIVS-II-16of(rs10768683)were
determinedbygenesequencing.Restrictionenzymesusedfor
thehaplotype analyseswere purchasedfrom New England
Biolabs,Inc.(Beverly,MA,USA).Classificationofhaplotypes
wasbasedonOrkinetal.21andNageletal.fortheSgene.22
Theassignmentofspecifichaplotypesinheterozygousstates
shouldbeinterpretedwithcautioniffamilystudiesorallele
cloning are notperformed. Inthe present study,the
inter-pretationoftheresultswasfacilitatedbyourpreviousreport
showingthatin206HbSSchildrenfromMinasGerais,98.5%
oftheSchromosomeswereofthetypesCARorBen.23
ACoulterT-890hematologycounterwasusedtoperform
all blood cell counts. Reticulocytes were counted in blood
smears stained with brilliant cresyl blue. All
hematologi-calvaluesweretranscribedfromthemedicalrecordsinthe
absence ofacuteclinical manifestationsand atleast three
monthsaftertheuse ofbloodproducts.Themathematical
averageofeachitemwasconsideredasthebaselinevaluefor
eachpatient.TherelativebaselineconcentrationofHbFwas
obtainedfromtheHbelectrophoresisresultsreportedineach
patient’smedicalrecord.Theresultsofelectrophoresis
test-ingperformedattheoldestagepossiblewithinthefollow-up
periodwere usedaslongasthesamplehadbeencollected
aftertwoyearsofage.
TranscranialDoppler(TCD)examinationswereperformed
in14childrenandinterpretedbyasingleexpertusinga
Nico-let equipment (modelEME TC 2000, Nicolet, Madison,WI,
USA).High-risk TCD wasdefinedasatime-averaged mean
ofthemaximumvelocity(TAMMX)≥200cm/sintheinternal
carotidormiddlecerebralarteryasoriginallydefinedbystroke
preventioninsicklecell anemia(STOP)investigators.24 The
examinationcouldnotbeperformedinfivepatients: three
examinationswereimpossiblebecauseofthelackof
cooper-ationonthepartofthechildrenandtwowerenotperformed
becausethechildrenfailedtoattendtheexamination.
Statisticalanalyseswere performedusing theStatistical
PackagefortheSocialSciencesprogram(SPSS),version20.0.
Quantitativeresultsareexpressedastheaverage±standard
deviationorasthemedianandrangewhendistributionwas
non-Gaussian.Prevalencewasexpressedasapercentageand
a95% confidenceinterval(CI)wasapplied.Unpaired ttests
wereusedtocomparemeanvaluesbetweenHbS/D-Punjab
andHbS-KorleBugroups.Testresultswereconsidered
signif-icantwhentheprobabilityofalphaerrorwas≤0.05.
ThestudywasapprovedbytheEthicsResearchCommittee
attheinvolvedinstitutions(caseNo.13327713.5.0000.5149).It
wasconductedinaccordancewiththeHelsinkiDeclarationas
revisedin2008.Patientsand/orguardianswereaskedtosign
theinformedconsentform.
Results
The incidence of Hb FSD at birth was 1:171,000 (95% CI:
1:120,000–1:299,000). Molecular analyses were applied to
samplesfrom19outofthe21childrenbecauseaspreviously
mentioned,samplecollectionfromtwochildrenwasnot
pos-sible.Theagesrangedfrom2.8to16.2years,withamedian
of8.9years.Thirteenchildrenweremale(68.4%)andsixwere
female(31.6%).
Outofthe19children,11children(tenfamilies)were
diag-nosedwithHbS/D-Punjabdiseaseandeightwerediagnosed
withHbS-KorleBu.Noothervariantswerefound.Gender
dis-tributionintheHbS/D-Punjabgroupwasfourfemales(36.4%)
andsevenmales(63.6%)andgenderdistributionintheHb
S-KorleBugroupwastwofemales(25%)andsixmales(75%).The
medianagewas11years(range:2.8–16.2)intheHbS/D-Punjab
groupand7.7years(range:3–13.7)intheHbS-KorleBugroup.
AllchildrenoftheHbS/D-Punjabgroupwerefoundtohave
baseline Hblevels below10g/dL(average: 8.0g/dL) and the
reticulocytecountsvaried.Thebaselinevaluesofthe
hema-tologic testsandgeneticresultsforeach childarereported
inTable1.Figure2comparesthemainresultsfoundinboth
groups. TheHbS/D-Punjabgroupwas foundtohave lower
average baseline Hb values and higher reticulocyte counts
thantheHbS-KorleBugroup(p-value<0.001forboth
com-parisons). Relative Hb Fconcentrations were higher in the
children ofthe HbS/D-Punjab group(p-value=0.001). Total
leukocyteandplateletcountswerealsohigherintheHb
S/D-Punjabgroup(p-value=0.003andp-value=0.06,respectively).
MeanratiosbetweenHbSandHbDconcentrationswere1.06
and 1.14 in the Hb S/D-Punjab and Hb S-KorleBu groups,
respectively(p-value=0.61).
The coinheritance of alpha-thalassemia (␣␣/−␣3.7) was
detected in four children (two with Hb S/D-Punjab and
two with Hb S-Korle Bu). Mean corpuscular volume (MCV)
and mean corpuscular Hb (MCH) were significantly lower
in children who co-inherited the alpha-thalassemia
dele-tion(p-value=0.008andp-value=0.03,respectively).TheHb
S/D-Punjabgroupwasanalyzedseparately:theMCVsofthe
patientswithandwithout-alphathalassemiaaveraged76.6fL
and85.9fL,respectively(p-value=0.001),whiletheMCHs
aver-aged24.2pgand29.1pg,respectively(p-value=0.001).
Intermsoftheassociatedclinicalfindings,two(18.2%)of
the childrenintheHb S/D-Punjabgrouphad acutesplenic
sequestrationcrises(ASSCs),andall11experiencedatleast
oneacutepainepisode.However,therewerenoASSCsinthe
HbS-KorleBugroup,andthreepatients(37.5%)hadatleast
oneacutepainepisodereportedassuchinthepatients’
med-icalrecords.NoneofthechildrenintheHbS-KorleBugroup
receivedbloodtransfusions;however,seven(63.6%)children
intheHbS/D-Punjabgroupreceivedtransfusions.
Nochildhadanovertstroke.MeanDopplerTAMMXvalues
forHbS/D-PunjabandHbS-KorleBuchildrenwere131.1cm/s
[standarderrorofthemean(SEM):6.7] and89.2cm/s(SEM:
6.5),respectively(p-value=0.001).Allchildrenwereclassified
asbeingatlowriskforstrokes.
BaselineoxygensaturationwaslowerintheHbS/D-Punjab
groupandsignificantlydifferedfromtheHbS-KorleBugroup.
Averagevaluesinthetwogroupswere91.9%and97%,
respec-tively (p-value=0.002). There were no deaths in the study
population.
Clinicaltreatmentwasalsoreviewed.Allpatientsreceived
aprescriptionforantimicrobialprophylaxis,dailyfolic acid
Table1–Meanbaselinehematologicaldataandgeneticresultsof11childrenwithHbS/DPunjabandeightchildrenwith HbS-Korle-Bu.
Id/Gender Hemoglobin
(g/dL)
Leukocytes (×109/L)
Platelets (×109/L)
Reticulocytes (%)
HbF(%) HbS(%) HbD(%) S
haplotype/␣−3.7 thalb
ChildrenwithHbS/DPunjab(n=11)
1/F 7.2 13.9 347.7 4.0 13 48 37 CAR/−
2/M 7.9 16.1 431.8 14.7 13 45 39 ATP/−
3/F 6.8 13.7 368.0 8.7 14 38 44 BEN/−
4/M 6.8 18.0 552.0 7.5 4 44 37 CAR/−
5/F 8.3 15.2 300.2 9.5 22 41 35 BEN/−
6/Ma 8.6 11.2 335.0 3.9 23 33 40 BEN/+
7/F 9.5 12.6 353.8 3.2 16 47 39 BEN/−
8/Ma 8.9 15.0 454.0 6.4 21 36 40 BEN/−
9/M 8.4 13.6 432.2 10.3 10 40 46 CAR/+
10/M 6.2 13.3 308.8 8.7 16 39 35 CAR/−
11/M 9.5 11.2 430.9 5.9 25 37 36 BEN/−
Mean 8.0 14.0 392.2 7.5 16.1 40.7 38.9
ChildrenwithHbS-Korle-Bu(n=8)
1/M 11.7 12.2 397.3 1.8 6 58 34 CAR/+
2/M 12.8 10.8 366.9 1.2 9 45 45 CAR/−
3/M 11.5 7.1 326.6 1.0 7 45 42 CAR/−
4/F 12.8 7.7 257.0 1.1 6 59 33 BEN/−
5/F 13.2 10.1 275.6 0.4 9 41 48 CAR/+
6/M 12.3 11.4 331.7 0.6 6 39 52 CAR/−
7/M 12.0 14.3 407.9 1.3 4 50 44 CAR/−
8/M 11.8 10.5 266.3 0.8 8 39 50 CAR/−
Mean 12.3 10.5 328.7 1.0 6.9 47.0 43.5
a Brothers.
b Co-inheritance(+or−)ofthealpha-thalassemiagene−␣3.7(−␣3.7/␣␣).
15.0
A
B
C
D
10.0
5.0
Mean hemoglobin concentr
ation (g/dL)
Mean f
etal hemoglobin, %
Mean leuk
ocyte count (x10E9/L)
Mean reticulocyte count, %
.0
25
20
10 15
0 5
20.0
15.0
5.0 10.0
0 15.0
10.0
5.0
.0 D-Punjab
P<.001
P=.001
P<.001
P=.003 Korle-Bu
D-Punjab Korle-Bu
D-Punjab Korle-Bu
D-Punjab Korle-Bu
Figure2–Meanbaselinehematologicaldataof11childrenwithHbS/D-PunjabandeightchildrenwithHbS-KorleBu.(A)
Totalhemoglobinconcentration(g/dL);(B)reticulocytes(%);(C)fetalhemoglobin(%);(D)leukocytecount(×109/L).The
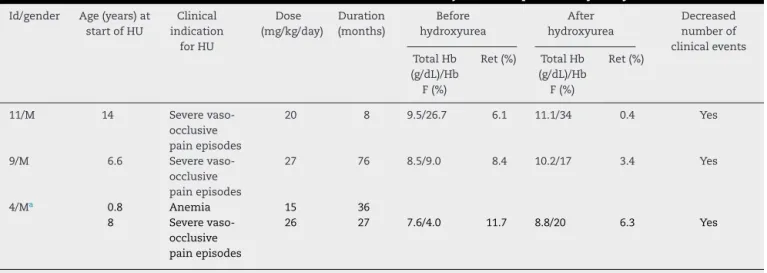
Table2–ClinicalandlaboratorialdatainthreechildrenwithHbSD-Punjabandresponsetohydroxyurea.
Id/gender Age(years)at startofHU
Clinical indication
forHU
Dose (mg/kg/day)
Duration (months)
Before hydroxyurea
After hydroxyurea
Decreased numberof clinicalevents TotalHb
(g/dL)/Hb F(%)
Ret(%) TotalHb (g/dL)/Hb F(%)
Ret(%)
11/M 14 Severe
vaso-occlusive painepisodes
20 8 9.5/26.7 6.1 11.1/34 0.4 Yes
9/M 6.6 Severe
vaso-occlusive painepisodes
27 76 8.5/9.0 8.4 10.2/17 3.4 Yes
4/Ma 0.8 Anemia 15 36
8 Severe
vaso-occlusive painepisodes
26 27 7.6/4.0 11.7 8.8/20 6.3 Yes
Id:identificationreferstonumbersinTable1;HU:hydroxyurea;Ret:reticulocytecount.
a ThischildwasgivenhydroxyureainAugust2005becauseofHb<5.0g/dL.HydroxyureawaswithdrawninJuly2008byhismother,notbythe
hematologist.ThedrugwasrestartedinSeptember2012,becauseofmultipleepisodesofvaso-occlusivepain.
childrenwithHbS/D-Punjabdiseasewerefoundtohaveused
hydroxyurea,asindicatedforrecurringepisodesofpain
cri-sis.AllchildrenpresentedincreasedtotalHbandHbFaswell
aslower reticulocyte countsand fewerpain episodes after
hydroxyureatherapy.Anotherpatientwhohadalsoreceived
anindicationfortheuseofhydroxyureabecauseofrecurring
paincrisesislikelytobeginusingthemedicationsoon.Table2
showsthedataontheuseofhydroxyurea.
HaplotypingofHbS/D-Punjabshowedthatallpatients
pre-sentedhaplotypeIfortheD-Punjaballele (+----+++,forthe
eightanalyzedpolymorphicsitesinthe5′->3′ direction).In
respecttothe-globingenecluster,sixwereBenin,fourCAR,
andonehadanatypicalhaplotype(+----+++,e.g.,haplotypeI
inhomozygosis).Similarly,allHbS-KorleBuallelespresented
haplotypeI.SevenhadtheSCARhaplotypeandonlyone,the
SBeninhaplotype.
Comparing hematological data between S CAR and S
Benin haplotypes withinthe Hb S/D-Punjab group of
chil-dren,Hbconcentration(7.2g/dLversus8.6g/dL,respectively)
andfetal Hb (10.8%versus20.2%,respectively)were
signif-icantly higher for the Benin haplotype (p-value=0.05 and
p-value=0.01,respectively).Nootherdata,includingDoppler
TAMMXvalues, oxygen saturation,andnumber of
transfu-sions, were significantlydifferent betweenCAR and Benin
groups.
Discussion
Two variants with the IEFprofile of Hb D were identified:
D-PunjabandKorleBu.Theclinicalandlaboratory
character-isticsofthetwogroupswereverydifferent.
Children with Hb S/D-Punjab disease presented many
different symptoms. This clinical courseis similar to that
observedinchildrenwithhomozygousHbSSdiseaseashas
alreadybeenreportedinotherstudies.10,11,16,17,25Oberoietal.
evaluatedten patientsagedbetween1and19 years.All of
them presentedwith moderate or severe anemia(average:
6.8g/dL)andatleastoneclinicalcomplicationrelatedtothe
disease,suchaspaincrisis,acutechestsyndrome,gallstones,
avascularnecrosisoffemoralhead,andrecurrentinfections.
Furthermore,eightpatientsrequiredanaverageofthreered
bloodcelltransfusionsduringtheirclinicalfollow-upperiod.11
In the present study, blood transfusions were required by
63.6%ofpatients.
Italiaetal.reportedon15patientswithagesbetween1and
34years.Theyclassifiedonlyonepatientasmild;twowere
classifiedashavingexperiencedmoderatecrises,and12were
foundtohaveseverehemolyticanemia;these12presented
recurrent pain crisesand requiredfrequent transfusions.10
El-Kalla & Mathews evaluated nine patients aged between
3and 10 yearsinthe UnitedArabEmirates. Theyreported
paincrisesofvaryingintensity, acutesplenicsequestration
crises,andrepeated infectionsinsevenpatients.Theother
twochildrenwereasymptomatic,althoughanemicandwith
increasedreticulocytecounts.17Thelargestsamplesizeinthe
literaturewaspublishedbyPateletal.Of42Indianpatients
with anaverage ageof22 years,25 were considered tobe
severe casesforpresentingthreeor moreacutepaincrises
and/ortheneedoftwoormorebloodtransfusionsintheyear
priortothestudy.25
ThegroupofchildrenwithHbS-KorleBudiseasedidnot
presentclinicalcomplications.Threechildrenpresentedwith
mildtomoderatediffuseabdominalpainthatwasconsidered
secondarytotheunderlyingdisease.However,apaincrisis
isfrequentlyinferredasthecauseofabdominalpainwhen
signsandsymptomsrelatedtootheretiologies(suchasfever,
vomiting,andbloodinthestools)areabsentinchildren.
Dur-ingthecrises,thesechildrenaregenerallyassumedtohaveHb
S/D-Punjabeveniftheyhavenotbeensubmittedtomolecular
testing.Paincrisesmaybedifficulttodistinguishfromother
equallyprevalentetiologiesinchildrensuchasconstipation
and functionalabdominalpain.26 Itisknown that
abdomi-nalpainaffects38%oftheschool-agedpediatricpopulation
weekly; noreliablebiologicalmarkersclearlydefine a
diag-nosisoffunctionalpain.27Inaddition,fewstudiesshowthat
theclinicalcourseofHbS-KorleBudiseasecarriersmaybe
ofpatientsandlengthoffollow-upprecludeasolid
conclu-sion.Someauthorshavecomparedthesepatientstosickle
celltraitcarriers.7Therefore,theexactdefinitionoftheHbD
variantthatthepatienthasisimportantforthecorrect
dif-ferentialdiagnosisofabdominalpaininchildren.Italsoaids
indecreasingboththe unnecessarystigmaagainstchronic
diseasesandunnecessarytreatments,suchasthe
indiscrim-inate use of powerful anti-inflammatory medications and
painkillers.
Thereseems tobe noreports availableinthe literature
abouttranscranialDopplerexaminationsinchildrenwithHb
SDdisease.Inthepresentstudy,allchildrenwerefoundtobe
atlowriskforstrokes.Asexpected,themeanvalueofTAMMX
wassignificantlyhigherinHbS/D-PunjabthaninHbS-Korle
BuchildrenbecausetheHbconcentrationwas significantly
lowerintheformergroup.
HigherHbFbaselinepercentageswereobservedintheHb
S/D-Punjabgroup;therewasastatisticallysignificant
differ-encefrom thevaluesobservedintheHbS-KorleBugroup.
Theinfluenceofthispercentageondecreasingsymptomsin
childrenwithHbS/D-Punjabdisease remainsunclear. This
associationhasbeen described inpatientswithHb SS
dis-easeandhasbeenexplainedbythefactthatredbloodcells
withlargerquantitiesofHb Fpossess lowerlevelsofHbS;
therefore,theyhavealowerchanceofsickling,andasaresult,
alowerprobabilityofexperiencingclinicalmanifestations.28
Patel et al. found higher Hb F concentrations to be
asso-ciated with a lower frequency of acute pain crises in the
42patientswithHbS/D-Punjabdisease.25 Meanwhile,three
other studies failed to find this association;however, they
did include much smaller sample populations than Patel
etal.Theotherstudiesincludedfive,nine,and15patients,
respectively.9,10,17
ThreechildrenwiththeHbS/D-Punjabvarianttook
hydrox-yurea, and all childrenexperiencedsignificant clinical and
laboratoryimprovements.InchildrenwithHbSS,theuseof
hydroxyureahasbeendeterminedtobesafeandtoprovide
satisfactory results.29 In patients with Hb SD disease, the
dataontheefficacyandsafetyofthismedicationarelimited
toisolated cases19 andtoafewstudieswithalarger
num-berofcases.Out oftheten childrenincludedinthe study
byOberoietal.,fivereceivedhydroxyureatotreatrecurrent
painepisodesand/orsevereanemia.Theirfollow-upperiods
rangedfrom6to50months.Inallcases,therewasadecrease
inthe number ofpaincrisesand alack ofsignificantside
effects.11Thelargestcohortincluded20Indianpatientsaged
between1and 45years who tookhydroxyurea foratleast
twoyears.Decreasedfrequenciesofcrisesandinterruptions
intransfusionswererecordedforallpatients.Nosideeffects
wereobserved.18
HaplotypeIisthecommonesttypeofD-Punjaballele in
almost all ethnic populationsso far described.6,14,15,30 The
exceptionisThailand,wherehaplotypeIIwasfoundinnine
patientsfromfivefamilies.31Soitisimpossibletotraceback
theoriginofthealleleinthepopulationofthisstudy.
Haplo-typesfortheHbKorleBuallelehavenotbeendescribedyet.
Becauseall childreninthepresent studyare alsotypeI, it
isimprobablethattheoriginoftheallelewillbedetermined
forsure.Itwouldbeinterestingtoknowwhetherthesame
haplotypeispresentinWesternAfricawherethemutation
seeminglyoriginated.5
Asfarasisknownfrominternationalreports,thepresent
studydemonstratedforthefirsttimethat,withintheHb
S/D-Punjabgroup,childrenwiththeSBeninhaplotypehadhigher
totalHbconcentrationsandhigherrelativeconcentrationsof
fetalHbthanchildrenwiththeSCARhaplotype.
Thelimitationsofthepresentstudyincludethefactthat
itwasaretrospectiveanalysisandalownumberofpatients
wereinvolved.Thesefactorslimitconclusionsregardingthe
incidenceofclinicalcomplicationsincasesofHbS/D-Punjab
andHbS-KorleBudiseaseaswellasareliableevaluationofthe
late-onseteffectsoftheuseofhydroxyurea.Furthermore,had
high performanceliquidchromatography(HPLC)been used
astheprimaryscreeningtestatPTN-MG,childrenwithHb
S-KorleBuwouldnotbeclassifiedashavingHbSD,butinstead
ashavinganunknownvariantwithintheHPLCwindowofHb
A2/HbE.
Inconclusion,thisisthefirststudyinBraziltoevaluate
childrenwithHbSDdiseasedetectedatbirthbyIEF.Itoffers
acleardescriptionoftwovariants:HbS/D-PunjabandHb
S-KorleBu.Earlynewbornscreeningandthesystematicgenetic
studyofHbDvariantsareusefulintreatingpatientsandin
informingthefamilymembers abouttheprognosisofeach
variant.ChildrenwithHb S/D-Punjabhaveaclinicalcourse
similartothosewithHbSSdisease.ThesubgroupwiththeS
CARhaplotypehavelowertotalHbconcentrationsandlower
relativeHbFconcentrationsthanthosewiththeSBenin
hap-lotype,butthelimitednumberofpatientsprecludesdefinitive
conclusions. In contrast,the clinical courseand laboratory
datainchildrenwithHb S-KorleBuappeartobesimilarto
thoseinchildrenwiththesicklecelltrait,butcautionshould
betakenwiththisstatement,consideringthelimitednumber
ofchildrenandshortfollow-upthusfarreported.
Funding
Fundac¸ãoHemominas,NewbornScreeningProgram
(Nupad-UFMG), Fundac¸ão de Amparo à Pesquisa de Minas Gerais
(FAPEMIG),andConselhoNacionaldeDesenvolvimento
Cien-tíficoeTecnológico(CNPq).
Conflicts
of
interest
Theauthorsdeclarenoconflictsofinterest.
Acknowledgments
Theauthorsacknowledgeallsubjectsand parentsfortheir
cooperationinthestudy.Theauthorsalsothankthe
finan-cial support of Fundac¸ão Hemominas, Newborn Screening
Program(Nupad-UFMG),Fundac¸ãodeAmparoàPesquisade
MinasGerais(FAPEMIG;grant#PPM-00780-15),andConselho
NacionaldeDesenvolvimentoCientíficoeTecnológico(CNPq;
r
e
f
e
r
e
n
c
e
s
1. SmithEW,ConleyCL.SicklecellhemoglobinDdisease.Ann InternMed.1959;50(1):94–105.
2. CaweinMJ,LappatEJ,BrangleRW,FarleyCH.HemoglobinS-D disease.AnnInternMed.1966;64(1):62–70.
3. TorresLS,OkumuraJV,SilvaDG,Bonini-DomingosCR. HemoglobinD-Punjab:origin,distributionandlaboratory diagnosis.RevBrasHematolHemoter.2015;37(2):120–6.
4. ItanoHA.Athirdabnormalhemoglobinassociatedwith hereditaryhemolyticanemia.ProcNatlAcadSciUSA. 1951;37(12):775–84.
5. HonigGR,SeelerRA,ShamsuddinM,VidaLN,MompointM, ValcourtE.HemoglobinKorleBuinaMexicanfamily. Hemoglobin.1983;7(2):185–9.
6. YavarianM,KarimiM,ParanF,NevenC,HarteveldCL, GiordanoPC.MulticentricoriginofHbD-Punjab [beta121(GH4)Glu→Gln,GAA>CAA].Hemoglobin. 2009;33(6):399–405.
7. AklPS,KutlarF,PatelN,SalisburyCL,LaneP,YoungAN. CompoundheterozygosityforhemoglobinS
[beta6(A3)Glu6Val]andhemoglobinKorleBu [beta73(E17)Asp73Asn].LabHematol.2009;15(3):20–4.
8. AdachiK,KimJ,BallasS,SurreyS,AsakuraT.Facilitationof HbSpolymerizationbythesubstitutionofGluforGlnatbeta 121.JBiolChem.1988;263(12):5607–10.
9. AdekileA,Mullah-AliA,AkarNA.Doeselevatedhemoglobin FmodulatethephenotypeinHbSD-LosAngeles?Acta Haematol.2010;123(3):135–9.
10.ItaliaK,UpadhyeD,DabkeP,KanganeH,ColacoS,SawantP, etal.ClinicalandhematologicalpresentationamongIndian patientswithcommonhemoglobinvariants.ClinChimActa. 2014;431:46–51.
11.OberoiS,DasR,TrehanA,AhluwaliaJ,BansalD,MalhotraP, etal.HbHbS/D-Punjab:clinicalandhematologicalprofileofa rarehemoglobinopathy.JPediatrHematolOncol.
2014;36(3):e140–4.
12.NagelRL,LinMJ,WitkowskaHE,FabryME,BestakM,Hirsch RE,etal.CompoundheterozygosityforhemoglobinCand KorleBu:moderatemicrocytichemolyticanemiaand accelerationofcrystalformation.Blood.1993;82(6):1907–12.
13.AdachiK,KimJ,KinneyTR,AsakuraT.Effectofthebeta73 aminoacidonthehydrophobicity,solubility,andthekinetics ofpolymerizationofdeoxyhemoglobinS.JBiolChem. 1987;262(22):10470–4.
14.PatelDK,MashonRS,PatelS,DashPM,DasBS.Beta-globin genehaplotypeslinkedwiththeHbD-Punjab
[beta121(GH4)Glu→Gln,GAA>CAA]mutationineasternIndia. Hemoglobin.2010;34(6):530–7.
15.RahimiZ,AkramipourR,KoraniS,NagelRL.HbD-Punjab [beta121(GH4)Glu→Gln]/beta0-thalassemia[IVSII.1(G→A)] intwocasesfromanIranianfamily:firstreport.AmJ Hematol.2006;81(4):302–3.
16.KelleherJFJr,ParkJO,KimHC,SchroederWA.
Life-threateningcomplicationsinachildwithhemoglobin SD-LosAngelesdisease.Hemoglobin.1984;8(3):203–13.
17.el-KallaS,MathewsAR.HbD-PunjabintheUnitedArab Emirates.Hemoglobin.1997;21(4):369–75.
18.PatelS,PurohitP,MashonRS,DehuryS,MeherS,SahooS, etal.Theeffectofhydroxyureaoncompoundheterozygotes forsicklecell-hemoglobinD-Punjab–asinglecentre experienceineasternIndia.PediatrBloodCancer. 2014;61(8):1341–6.
19.UddenMM,LoMN,SearsDA.Successfulhydroxyurea treatmentofapatientwithSDhemoglobinopathy.AmJ Hematol.1999;60(1):84–5.
20.TanAS,QuahTC,LowPS,ChongSS.Arapidandreliable 7-deletionmultiplexpolymerasechainreactionassayfor alpha-thalassemia.Blood.2001;98(1):250–1.
21.OrkinSH,KazazianHHJr,AntonarakisSE,GoffSC,BoehmCD, SextonJP,etal.Linkageofbeta-thalassaemiamutationsand beta-globingenepolymorphismswithDNApolymorphisms inhumanbeta-globingenecluster.Nature.
1982;296(5858):627–31.
22.NagelRL,FabryME,PagnierJ,ZohounI,WajcmanH,BaudinV, etal.Hematologicallyandgeneticallydistinctformsofsickle cellanemiainAfrica.TheSenegaltypeandtheBenintype.N EnglJMed.1985;312(14):880–4.
23.BelisárioAR,MartinsML,BritoAM,RodriguesCV,SilvaCM, VianaMB.-Globingeneclusterhaplotypesinacohortof221 childrenwithsicklecellanemiaorS◦-thalassemiaandtheir
associationwithclinicalandhematologicalfeatures.Acta Haematol.2010;124(3):162–70.
24.AdamsRJ,McKieVC,HsuL,FilesB,VichinskyE,PegelowC, etal.Preventionofafirststrokebytransfusionsinchildren withsicklecellanemiaandabnormalresultsontranscranial Dopplerultrasonography.NEnglJMed.1998;339(1):5–11.
25.PatelDK,PurohitP,DehuryS,DasP,DuttaA,MeherS,etal. Fetalhemoglobinandalphathalassemiamodulatethe phenotypicexpressionofHbHbS/D-Punjab.IntJLab Hematol.2014;36(4):444–50.
26.RhodesMM,BatesDG,AndrewsT,AdkinsL,ThorntonJ, DenhamJM.Abdominalpaininchildrenwithsicklecell disease.JClinGastroenterol.2014;48(2):99–105.
27.SapsM,SeshadriR,SztainbergM,SchafferG,MarshallBM,Di LorenzoC.Aprospectiveschool-basedstudyofabdominal painandothercommonsomaticcomplaintsinchildren.J Pediatr.2009;154(3):322–6.
28.SteinbergMH,RodgersGP.Pharmacologicmodulationoffetal hemoglobin.Medicine.2001;80(5):328–44.
29.ThornburgCD,FilesBA,LuoZ,MillerST,KalpatthiR,IyerR, etal.ImpactofhydroxyureaonclinicaleventsintheBABY HUGtrial.Blood.2012;120(22):4304–10.
30.AtalayEÖ,AtalayA,ÜstelE,YildizS,OztürkO,KöselerA, etal.GeneticoriginofHbD-LosAngeles[121(GH4)Glu→Gln, GAA→CAA]accordingtothe-globingenecluster
haplotypes.Hemoglobin.2007;31(3):387–91.
31.FucharoenS,ChangtrakunY,SurapotS,FucharoenG, SanchaisuriyaK.MolecularcharacterizationofHbD-Punjab [beta121(GH4)Glu→Gln]inThailand.Hemoglobin.