Une étude de la théorie fonctionnelle de la densité des propriétés électroniques et magnétiques des complexes de fer. La thèse présentée dans ce manuscrit est consacrée à l'étude des propriétés spectroscopiques et magnétiques de composés bioinorganiques à base de fer en utilisant la théorie fonctionnelle de la densité.
Généralités
Hamiltonien Moléculaire
Au début du XXe siècle, les physiciens découvrent que les lois de la mécanique classique ne permettent pas de décrire le comportement des petites particules comme les électrons [1], les noyaux ou les molécules. Celles-ci sont en effet régies par les lois de la mécanique quantique qui permettront de calculer et de prédire les propriétés physiques et chimiques des systèmes atomiques et moléculaires.
L’Approximation Born-Oppenheimer
4 Chapitre I Chimie théorique : concepts et méthodes Le terme défini en 1.A.4 ne joue aucun rôle dans le calcul des grandeurs physiques et n'est généralement pas pris en compte. Il est donc nécessaire de mettre en œuvre des procédures de simplification associées à quelques astuces mathématiques pour permettre d'obtenir une solution approchée.
L’Approximation Hartree-Fock
L’énergie de la fonction d’onde exacte peut ainsi servir de limite inférieure à l’énergie calculée pour toute autre fonction d’onde antisymétrique normalisée. A partir de la fonction d'onde définie en 1.A.6 on arrive à des équations mono-électroniques sous forme d'orbitales.
Fonctions de base
Le deuxième formalisme concerne les systèmes à couches dites « ouvertes » et consiste à traiter indépendamment les orbitales de spin α et β. On peut également privilégier les orbitales de valence à bases Split-Valence – SV – qui sont construites en augmentant uniquement le nombre de fonctions de la coque de valence et en ne gardant qu'une fonction de chaque type de symétrie pour les plans cardiaques.
Formulation de la Corrélation Electronique
10 Chapitre I Chimie théorique : concepts et méthodes Par exemple, une base DZ est construite en doublant le nombre de fonctions de base minimales pour décrire la fonction d'onde avec plus de flexibilité et de précision. En fin de compte, l’ajout de fonctions de polarisation augmentera la qualité des résultats ; ces fonctions décrivant la distorsion du nuage électronique par rapport à la symétrie sphérique de l'atome.
Les Méthodes Post-Hartree-Fock
- La Méthode Perturbative Møller-Plesset
- Les Méthodes Multi-Configurationnelles
- i L’Interaction de Configurations
- ii La Méthode de l’Espace Actif Complet : CASSCF
- iii La Méthode mixte CASPT2
Il s'agit d'une méthode développée à partir du modèle RHF qui permet d'obtenir une bonne approximation de la fonction d'onde d'ordre zéro dans des systèmes à niveaux d'énergie quasi-dégénérés. L'aspect monodéminantal permet une interprétation « chimique » de la fonction d'onde issue de ce type de formalisme.
Cadre de la Théorie de la Fonctionnelle de la Densité
- La Densité Electronique
- Premier théorème de Hohenberg-Kohn
- Deuxième théorème de Hohenberg-Kohn
- Equations de Kohn-Sham : Approche orbitalaire
Rappelons que pour un système électronique décrit par l'hamiltonien H (équation 1.B.17), l'énergie et la fonction d'onde de l'état fondamental sont déterminées par la minimisation de la fonctionnelle E[ ]ψ. L'énergie d'un système de N électrons en interaction est donc fonction de la densité, et la recherche de l'énergie de l'état fondamental peut se faire de manière itérative sur la base d'une loi variationnelle.
Traitement de l’échange et de la corrélation
- Justification de l’approche de Hohenberg et Kohn
- Approximation locale
- L’approximation du gradient généralisé
- Les fonctionnelles hybrides
- Les logiciels
La fonction d'échange B88 est basée sur une analyse dimensionnelle et sur un comportement asymptotique correct de la densité d'énergie d'échange. Cette fonction est basée sur une analyse de l'expansion graduelle du trou d'échange-corrélation autour de sa forme LSDA.
Les applications de la DFT en chimie bio-inorganique
Les méthodes DFT permettent une bonne estimation de la structure des systèmes bio-inorganiques. Ainsi, le meilleur choix pour ce type de calcul semble être celui des fonctionnelles hybrides sachant par ailleurs que l'écart qui existe entre les états haut spin et bas spin est lui-même fonction de la fraction d'échange introduite au sein de la fonctionnelle utilisée. 43].
L’atome de fer dans tous ses états
Niveaux d’énergie de l’ion libre
- Hamiltonien électronique
- Configuration d n et microétats
- Répulsion électronique et termes spectroscopiques
- Couplage spin-orbite
- Polarisation de spin
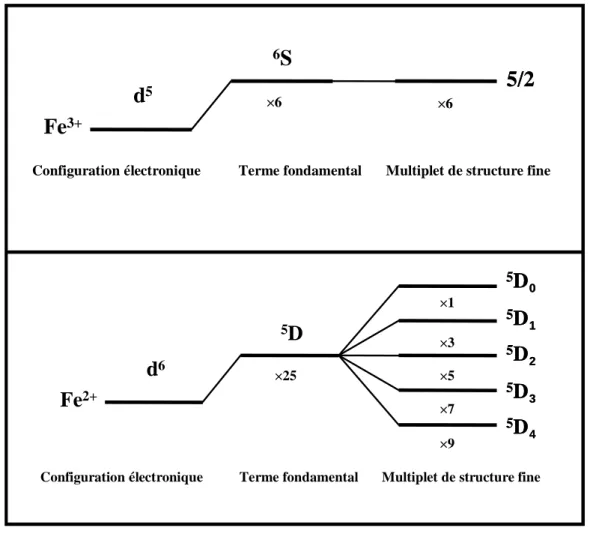
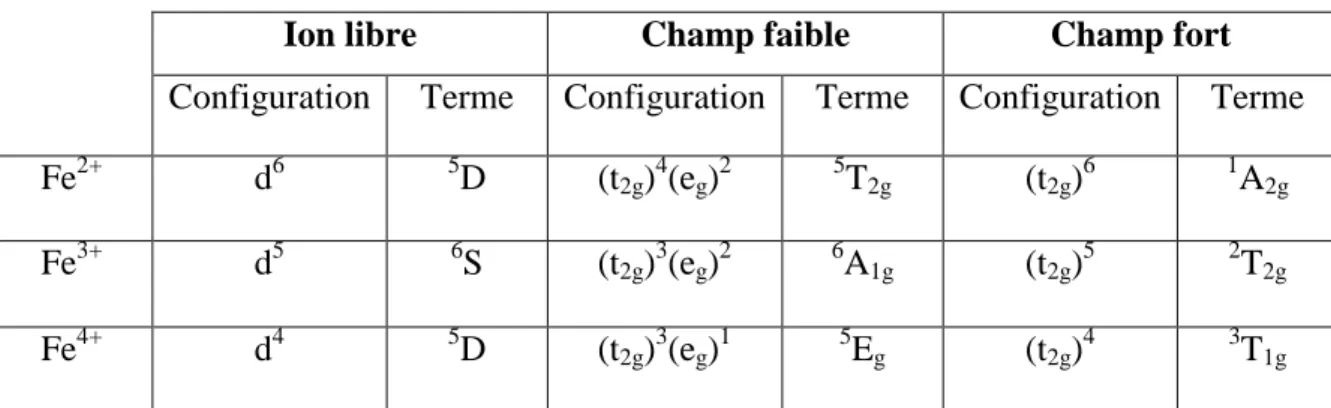
La mécanique quantique permet d'obtenir les niveaux d'énergie d'une entité chimique en calculant les valeurs propres de l'hamiltonien correspondant. Cet état fondamental dans le cas de l’ion fer Fe2+ de configuration électronique d6 est donc 5D4.
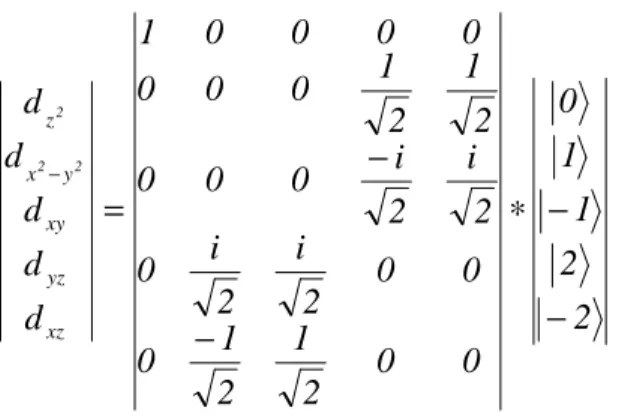
L’ion de fer et son environnement
- Levée de dégénérescence du niveau 3d
- i Symétrie sphérique
- ii Symétrie octaédrique
- iii Symétrie tétraédrique
- iv Termes spectroscopiques
- Champ fort versus champ faible
- Effet Jahn-Teller et abaissement de symétrie
- Blocage du moment cinétique orbital
46 Chapitre II L'atome de fer dans tous ses états Un traitement complet consiste à inclure en outre les niveaux d'énergie des molécules. 54 Chapitre II L'atome de fer dans tous ses états En général, selon la configuration électronique, c'est possible. Dans ce qui suit, nous décrirons l’ion fer dans un complexe en utilisant la théorie des champs de ligands afin de pouvoir calculer la covalence de la liaison métal-ligand.
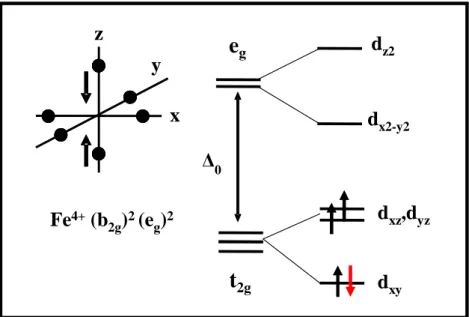
60 Chapitre II L'atome de fer dans tous ses états Dans le cas du complexe [(NH3)5FeIVO]2+, représenté sur la Figure 2.16, le moment orbital de.
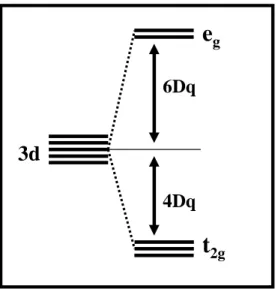
Magnétisme et Interaction d'échange
L'interaction d'échange isotrope
- Introduction
- Premiers développements
- L'hamiltonien effectif HDVV
- Les différentes approches
- i L'approche"Liaison de Valence"
- I.ii L'approche "Orbitale Moléculaire"
- iii L'approche "Symétrie Brisée"
A partir de ces deux configurations, on définit la constante d'échange J comme la différence d'énergie orbitale entre les états de spin S = 1 et S = 0. La constante d'échange sera négative dans le cas d'un couplage antiferromagnétique et l'état électronique fondamental sera alors l'état de spin minimal S = Sa −Sb. Il est associé au moment de spin total M = 0 et n'est pas un état monodéterminant.
82 Chapitre III Magnétisme et interaction d'échange Pour l'état de spin pur S = 0 qui nous intéresse, on sait qu'il résulte d'un couplage antiparallèle des spins locaux de chaque ion fer.
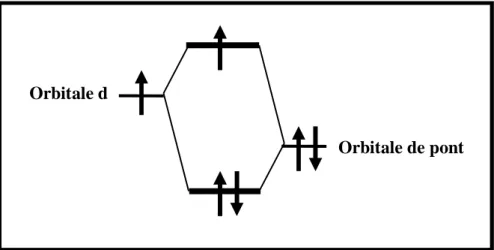
La délocalisation électronique
- L' interaction de double échange
- i Cas général
- ii En l'absence d'asymétrie locale
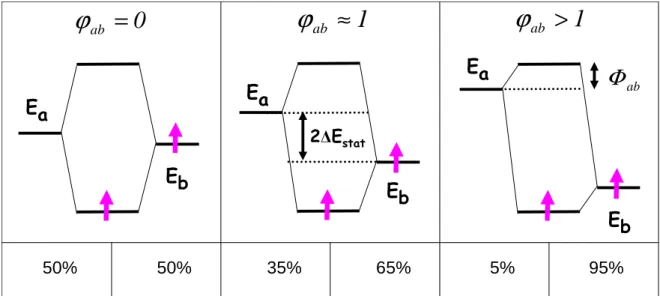
86 Chapitre III Magnétisme et interaction d'échange Appliquons cette classification au cas des dimères fer-soufre de valence mixte. Dans ces conditions, nous déterminerons les effets respectifs des termes d'échange isotrope JHeis et d'échange double B par rapport à la localisation de l'électron supplémentaire au sein d'un dimère à valence mixte. B ≠ 0 et Jab >0 : Le cas général des systèmes fer-soufre On aura une concurrence entre les deux termes.
Aux faibles valeurs du rapport B/Jab, le couplage antiferromagnétique domine et l'état électronique fondamental du système a un spin total de S=1/2 : l'interaction d'échange isotrope domine l'interaction d'échange double.
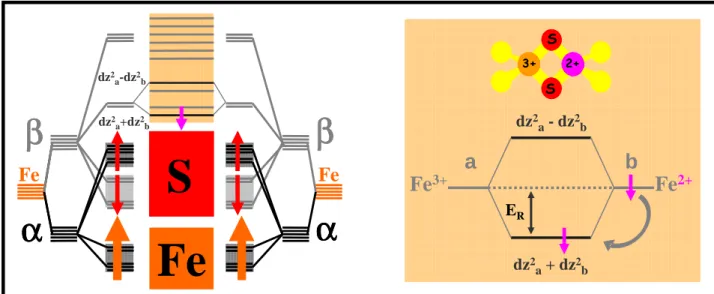
La localisation électronique
- La contribution statique…
- La contribution dynamique
- i Introduction
- ii Couplage vibronique et interaction de double échange
- Couplage des effets dynamiques et statiques
136 Chapitre IV Spectroscopique et Hamiltonien de Spin Nous pouvons résumer tous les cas essentiels comme suit. Les calculs de la densité électronique totale ρeA( )0 et du burst quadripolaire ∆EQcalc ont été réalisés à l'aide du logiciel de chimie quantique ADF 2006 en précision six et avec une base Triple Zeta plus deux fonctions de polarisation (TZ2P). Les disques colorés représentent ici le cycle porphyrine et (X) est un ligand variable correspondant à une tyrosine (ligand oxygéné) dans le cas de la catalase.
Ces investigations, réalisées par spectroscopie d'absorption des rayons X (EXAFS) et par la technique de Mössbauer, ont montré qu'à pH acide, le composé II du système CPO était probablement un complexe ferryl protoné [12].
Spectroscopies et Hamiltonien de spin
Hamiltonien de spin
Lorsqu'on envisage une étude utilisant des techniques telles que la résonance paramagnétique électronique ou la spectroscopie Mössbauer, il est alors possible de décrire les systèmes susmentionnés à l'aide d'un hamiltonien phénoménologique fictif ou d'un hamiltonien Hsp-spin. Hél est l'hamiltonien électronique, HCF est le terme du champ cristallin et HLS est l'interaction spin-orbite. Signification et origine des termes de l'hamiltonien de spin L'hamiltonien de spin est défini comme suit.
Ce terme trouve sa source dans l’interaction spin-orbite, qui donnera également lieu à une contribution à l’interaction de bursting en champ nul D~SO.

La spectroscopie RPE
- Introduction
- L'effet Zeeman électronique
- Dérivation des grandeurs caractéristiques
- Tenseur g ~ , Eclatement à champ nul et Applications
- Autres contributions
- i L'éclatement à champ nul : origine spin-spin
- ii L'éclatement à champ nul : contribution anisotrope
- iii Terme d'échange antisymétrique
- iv Terme de double échange antisymétrique
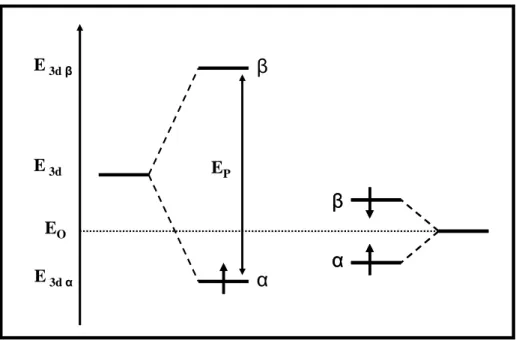
128 Chapitre IV Spectroscopies et hamiltonien de spin Le terme H est l'hamiltonien de perturbation qui rassemble le terme d'effet Zeeman p approprié et l'interaction spin-orbite telle que. Dans un cas très général le tenseur g~ est une matrice carrée symétrique d'ordre deux que l'on peut écrire comme suit. 124 Chapitre IV Spectroscopies et hamiltonien de spin Pour le cas du dimère fer-soufre à valence mixte, nous obtenons.
128 Chapitre IV Spectroscopies et hamiltonien de spin En pratique, l'interaction antisymétrique est à l'origine d'un ferromagnétisme faible, notamment dans le cas de réseaux étendus de faible symétrie.
La spectroscopie Mössbauer
- Introduction
- L'effet Mössbauer
- Hamiltonien de spin
- Grandeurs caractéristiques
- i Le déplacement isomérique
- ii L'éclatement quadripolaire
- iii Structure magnétique
L'ED associé à l'effet Doppler atteint une valeur optimale comparable à deux fois la valeur de l'énergie de retour ER, comme le montre le cas (b) de la Figure 4.12. Une autre méthode appelée « sans rebond » a été développée par Rudolf Mössbauer au cours de sa thèse, comme le montre l'exemple (c) de la figure 4.12. 134 Chapitre IV Spectroscopies et hamiltonien de spin La condition de résonance implique un changement dans la fréquence des rayons γ émis et reçus, donnant lieu à l'effet Doppler comme vu ci-dessus, par ex.
Étant donné que le rayon du noyau est différent selon qu'il est dans l'état fondamental d'énergie E ou dans l'état excité d'énergie g' E, le déplacement énergétique de la transition nucléaire e' est défini comme .
Méthodologie
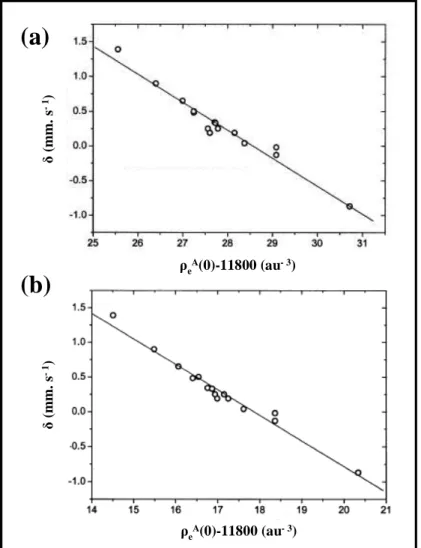
148 Chapitre V Spectroscopie Mössbauer et modélisation DFT Les quantités ρe( )r et ρs( )r sont accessibles par la théorie des orbitales moléculaires. MO), afin qu'ils puissent être modélisés par des calculs de théorie fonctionnelle de la densité (DFT), et nous allons maintenant présenter la procédure que nous avons suivie à cet effet. Étant donné que tous les facteurs formant l’expression 5.2 sont constants à l’exception de la quantité ρeA( )0, nous pouvons établir la connexion simplifiée suivante. Modéliser théoriquement la grandeur ∆EQ revient donc in fine à calculer les différentes composantes du tenseur EFG et à extraire ces trois valeurs propres.
Pour une orbitale moléculaire donnée ϕ0 correspondant à une combinaison linéaire des orbitales d de l'ion fer, les composantes Vij du tenseur EFG peuvent également s'écrire comme suit [2].
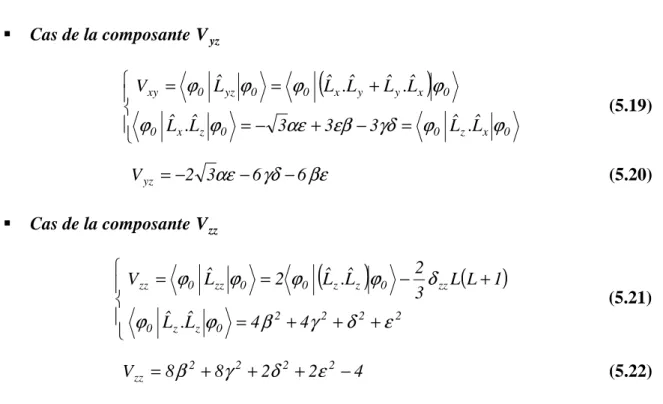
Le système de référence : le complexe de fer
- Etat de l'art
- Courbes de calibration
- i Modélisation du déplacement isomérique
- ii Modélisation de l'éclatement quadripolaire
- iii Conclusions
Pour établir la première corrélation entre calcul et expérience, nous avons testé la méthode DFT pure et la fonctionnelle d'échange-corrélation (GGA) BP86. Enfin, nous avons essayé une méthode hybride de DFT et fonctionnelle (B3LYP). Pour cela, nous avons utilisé la méthode DFT pure et testé la fonctionnalité pure du PW91.
Concernant la première partie de cette étude et la modélisation du déplacement expérimental des isomères par le calcul de la densité électronique totale, les résultats que nous avons obtenus sont convaincants.
Le système biologique : la catalase
- Etat de l'art
- i Présentation du composé
- ii Problématique du sujet
- Corrélations expérience/calculs théoriques
- i Etude de l'enzyme native
- ii Etude du composé II
- iii Etude du composé I
- iv Conclusions
172 Chapitre V Spectroscopie Mössbauer et modélisation DFT La première étape de la réaction catalase correspond à l'oxydation à deux électrons de l'ion. Pour démontrer la protonation du groupe ferryl dans les intermédiaires de la catalase, nous avons réalisé une étude visant à caractériser le composé II de la catalase de Proteus mirabilis (PMC). Dans un premier temps, la caractérisation du composé II issu des PMC a été réalisée par spectroscopie Mössbauer à bas champ [24], qui a permis de démontrer la coexistence de deux formes stables dépendantes du pH : une première à faible pH (LpH II) et une seconde à pH élevé (HpH II).
La première corrélation expérience-calcul que nous avons établie concernait la forme native de la catalase PMC.
Le système biologique : la Protéine Fer-Soufre
- Introduction générale : les centres [Fe-S] et [2Fe-2S]
- i Structures et Classification
- ii Rôle et Fonction
- Les différentes familles de protéines à centres [Fe-S]
- i Les Rubrédoxines
- ii Les autres familles : Desulfoférrodoxine et Rubrérythine
- Les différentes familles de protéines à centres [2Fe-2S]
- i Les Ferrédoxines
- ii Les Xanthine Oxydases
- iii Les protéines Rieske
- Les différentes familles de protéines fer-soufre complexes
- i La Nitrogénase
- ii L'Hydrogénase
- iii La Sulfite Réductase
- Analogues Synthétiques de protéines à centres fer-soufre
Modélisation du tenseur g ~ : une revue critique
- Etat de l'art et Modèles
- Etude des centres fer-soufre des protéines Rieske
- i Corrélations magnéto-structurales
- ii Limites du modèle de Bertrand et al
- iii Paramétrisation du calcul de tenseur g ~
Tenseur g ~ : nouvelle approche phénoménologique
- Compétition des termes ∆E et B et Matrice énergie
- La prise en compte du couplage vibronique
- i Premier cas : Β = 0 et ∆ E stat = 0
- ii Second cas : Β ≠ 0 et ∆ E stat ≈ 0
- iii Troisième cas : Β ≠ 0 et ∆ E stat ≠ 0
- Le calcul de la constante d'échange
- Le calcul du tenseur g ~
Applications aux protéines à centres [2Fe-2S]
- Présentation des systèmes étudiés
- Méthodologie DFT
- Ferrédoxines et Rieske
- Mutants de Ferrédoxines, de Rieske et Complexes Modèles
- Rieske déprotonées et Xanthines Oxydases
- Thiorédoxine
- Conclusions et Perspectives



![Figure 2.13 : Niveaux d'énergies du complexe fer-soufre [Fe II (SH) 4 ] 2-](https://thumb-eu.123doks.com/thumbv2/1bibliocom/464041.69645/75.892.163.654.707.1100/figure-niveaux-énergies-complexe-fer-soufre-fe-ii.webp)
![Figure 2.12 : Niveaux d'énergies du complexe fer-soufre [Fe III (SH) 4 ] 2-](https://thumb-eu.123doks.com/thumbv2/1bibliocom/464041.69645/75.892.214.701.128.517/figure-niveaux-énergies-complexe-fer-soufre-fe-iii.webp)
![Figure 2.14 : Schéma simplifié des niveaux d'énergies du complexe [(NH 3 ) 5 Fe IV O] 2+](https://thumb-eu.123doks.com/thumbv2/1bibliocom/464041.69645/76.892.202.732.440.816/figure-schéma-simplifié-niveaux-énergies-complexe-nh-fe.webp)