http://dx.doi.org/10.1590/s2175-97902017000300251
A
r
*Correspondence: C. G. Magalhães. Departamento de Química, Centro de Ciências Exatas e Naturais, Universidade Estadual de Ponta Grossa, 84.030-900, Ponta Grossa, Paraná, Brasil. Phone: +55 - 42 - 3220-3062. E-mail: [email protected]
Lupeol and its esters: NMR, powder XRD data and
in vitro
evaluation of cancer cell growth
Aline Teixeira Maciel e Silva
1, Cássia Gonçalves Magalhães*
2, Lucienir Pains Duarte
3, Wagner da
Nova Mussel
3, Ana Lucia Tasca Gois Ruiz
4, Larissa Shiozawa
4, João Ernesto de Carvalho
4,5, Izabel
Cristina Trindade
6, Sidney Augusto Vieira Filho
61Departamento de Produtos Farmacêuticos, Faculdade de Farmácia, Universidade Federal de Minas Gerais, Belo Horizonte,
Minas Gerais, Brasil, 2Departamento de Química, Centro de Ciências Exatas e Naturais,Universidade Estadual de Ponta
Grossa, Ponta Grossa, Paraná, Brasil,3Departamento de Química, Instituto de Ciências Exatas, Universidade Federal de
Minas Gerais, Belo Horizonte, Minas Gerais, Brasil, 4Centro Pluridisciplinar de Pesquisas Químicas, Biológicas e Agrícolas,
Universidade Estadual de Campinas, Paulínia, São Paulo, Brasil, 5Faculdade de Ciências Farmacêuticas, Universidade
Estadual de Campinas, Campinas, São Paulo, Brasil, 6Departamento de Farmácia, Escola de Farmácia, Universidade Federal
de Ouro Preto, Ouro Preto, Minas Gerais, Brazil
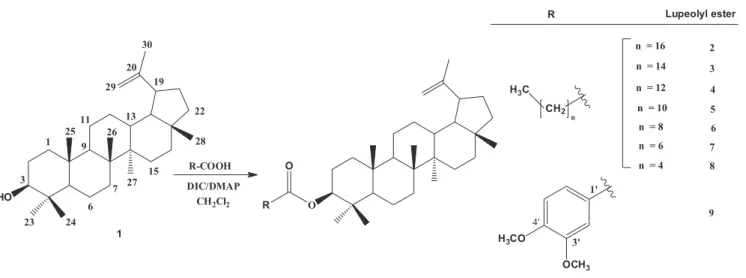
The triterpene lupeol (1) and some of its esters are secondary metabolites produced by species of Celastraceae family, which have being associated with cytotoxic activity. We report herein the isolation of 1, the semi-synthesis of eight lupeol esters and the evaluation of their in vitro activity against nine strains of cancer cells. The reaction of carboxylic acids with 1 and DIC/DMAP was used to obtain lupeol stearate (2), lupeol palmitate (3) lupeol miristate (4), and the new esters lupeol laurate (5), lupeol caprate (6), lupeol caprilate (7), lupeol caproate (8) and lupeol 3’,4’-dimethoxybenzoate (9), with high yields. Compounds 1-9 were identiied using FT-IR, 1H, 13C-NMR, CHN analysis and XRD data and were tested in vitro for proliferation of human cancer cell activity. In these assays, lupeol was inactive (GI50> 250µg/ mL) while lupeol esters 2 -4 and 7 - 9 showed a cytostatic efect. The XRD method was a suitable tool to determine the structure of lupeol and its esters in solid state. Compound 3 showed a selective growth inhibition efect on erythromyeloblastoid leukemia (K-562) cells in a concentration-dependent way. Lupeol esters 4 and 9 showed a selective cytostatic efect with low GI50 values representing promising prototypes for the development of new anticancer drugs.
Keywords: Lupeol/in vitro evaluation. Lupeol ester. K-562 cells. XRD method. Antiproliferative efect.
INTRODUCTION
Despite the efforts to develop new strategies of cancer prevention and therapy (Galmarini, Galmarini, Galmarini, 2012), cancers still represent a worldwide problem of public health. According to the Word Health Organization (Ferlay et al., 2012), around 14 million of
the new cancer cases occurred in 2012 (57% of this total in less developed regions). Among the strategies to treat cancer, cancer chemotherapeutic agents represent crucial tools basically aiming to eliminate or at least inhibit
tumor cell growth (Galmarini, Galmarini, Galmarini, 2012; Chabner, Roberts, 2005). Considering all antitumor chemotherapeutic arsenal approved between 1940s and 2014, 49% (85 chemical entities) were natural products per si or directly derived from them (Newman,
Cragg, 2016).
Among natural products, triterpenoids have been considered a promising class for cancer chemoprevention and chemotherapy (Salminen et al., 2008; Lachance et al., 2012; Gali-Muhtasib et al.,2015), and they have
been highlighted as antineoplastic agents (DallaVechia, Gnoatto, Gosmann, 2009; Laszczyk, 2009; Siddique, Saleem, 2011; Sultana, 2011).
anti-inflammatory, antioxidant and antiangiogenic effects (Laszczyk, 2009; Siddique, Saleem, 2011). Lupeol (1, Figure 1), a pentacyclic triterpene, occurs in many
medicinal plants(Laszczyk, 2009), such as in leaves of Maytenus salicifolia Reissek (Celastraceae) (Núñez et al.,
2005). This compound has displayed anti-inlammatory property (Salminen et al., 2008; Saleem, 2009; Shahlaei et al., 2013),protective efect during low-density lipoprotein
(LDL) oxidation (Geetha, Varalakshmiu, Latha, 1998; Andrikopoulos et al., 2003), and anticancer activity
against different cell lines [melanoma (G361, 451Lu and WM35), T-lymphoblastic leukemia (CEM), breast carcinoma (MCF-7 and MDA-MB-231), lung carcinoma (A-549), multiple myeloma (RPMI 8226) and cervical carcinoma (HeLa)] (Saleem, 2009; Saleem et al.; 2008;
Gallo, Sarachine, 2009).
Some natural lupeol esters also present promising
biological efects such as antimalarial (Fotieet al., 2006)
and acetylcholinesterase inhibitory activities (Gurovic
et al., 2010). Based on these promising activities, some
lupeol esters have been synthetized and evaluated for different activities (Li et al., 2013; Lachance et al.,
2012; Reddy et al., 2009; Sudhahar, Kumar, Varalaksmi,
2006a). For example, lupeol linoleate has been described as efective to reduce hypercholesterolemia (Sudhahar, Kumar, Varalaksmi, 2006a; Sudhahar, Kumar, Varalaksmi, 2006b; Sudhahar et al., 2007a)and also as a protective
agent in diferent oxidative stress conditions (Sudhahar, Kumar, Varalaksmi, 2006a; Sunitha, Nagaraj, Varalaksmi, 2001; Sudhahar et al., 2007b; Sudhahar et al., 2008;
Sudhahar, Veena, Varalaksmi, 2008).
The aim of this work was the semi-synthesis of eight lupeol esters, from which five (5 to 9) are new
ones. The compounds were characterized by Fourier transform infrared (FT-IR), nuclear magnetic resonance
(1H and 13C NMR) spectroscopy, CHN analysis and
powder X-ray difractometry (XRD). Moreover, lupeol and the eight lupeol esters were evaluated regarding their antiproliferative in vitro potential against a human cell lines panel.
RESULTS AND DISCUSSION
Synthesis and identification of lupeol esters
Lupeol (1) was isolated from hexane branch extract of M. salicifolia through phytochemical processes as
described in the literature (Magalhães et al., 2011). The esters 2 to 9 were obtained reacting 1 with an adequate
carboxylic acid and the DIC/DMAP reagents (Figure 1), with yields ranging from 86 to 96%.The 1H and 13C NMR
chemical shift assignments of compound 1 (S3) were in
accordance with the spectral data published by Shahlaei and coworkers (2013).
The structures of lupeol esters (2 to 9) were
conirmed due to the disappearance of signal at δC 71.0, in the 13C NMR spectra ( S4 to S11), corresponding to carbon
3 bonds in the hydroxyl group, the presence of signal at ~
δC 80.0 associated to C-O-C, together the of the signal at
~ δC 171.0 (C=O). The signal associated to C=O group to
compound 9 appeared at δC 166.10 due to the inluence of
3’,4’-dimethoxybenzoate group (Mahato, Kundu, 1994). The physical chemical data (IR, 1H and 13C NMR and
CHN analysis) of compounds 1 to 9 are described below,
as well as the amount obtained (mmol) and percent yield
for the esters 2 to 9.
3β-Lup-20(29)-en-3-ol (lupeol) (1): 426 g mol-1 [mp 213.8
- 215.2 ºC].
IR (KBr, cm-1): 3550, 3400, 3295, 2920, 2850, 1640
(weak) 1470, 1455, 1440, 1380, 1360, 1140, 1110, 1040, 985, 880.
1H NMR (CDCl
3, 200 MHz) δ:4.57 (s, H-29a), 4.68(s,
H-29b), 3.21(dd, J= 2.0; 6.0 Hz, H-3) ,1.68, 1.00, 0.97,
0.95, 0.83, 0.79, 0.76 (21 H, 7 s, 7 CH3).
13C NMR (CDCl
3, 50 MHz) δ: 38.08 (C-1), 27.43(C-2),
79.05 (C-3), 38.73 (C-4), 55.33 (C-5), 18.34 (C-6), 34.31 (C-7), 40.86 (C-8), 50.47 (C-9), 37.20 (C-10), 20.95 (C-11), 25.17 (C-12), 38.88 (C-13), 42.86 (C-14), 27.47 (C-15), 35.61 (C-16), 43.02 (C-17), 48.34 (C-18), 48.00 (C-19), 150.98 (C-20), 29.87 (C-21), 40.02 (C-22), 28.00 (C-23), 15.37 (C-24), 16.13 (C-25), 16.00 (C-26), 14.57 (C-27), 18.02 (C-28), 109.33 (C-29), 19.32 (C-30). CHN analysis: Calcd for C30H50O: C, 84.44; H, 11.81%.
Found: C, 84.49; H, 11.03%.
Lupeol stearate (2): 692 g mol-1, [0.85 mmol (86% yield)],
(mp 51.7 - 52.7 °C).
IR (KBr, cm-1): 3071, 2915, 2850 (CH), 1727 (C=O), 1640,
1172 (CO-O-C), 882.
1H NMR (CDCl
3, 200 MHz) δ:4.68(s, H-29b), 4.58 (s,
H-29a), 4.49 ( dd, J= 4.0; 8.0 Hz, H-3), 1.68, 1.06, 0.94,
0.90, 0.88, 0.83, 0.79 ( 21H, 7 s, 7 CH3).
13C NMR (CDCl
3, 50 MHz) δ: 38.40 (CH2, C-1), 23.74
(CH2, C-2), 80.61 (C,C-3), 37.83 (C,C-4), 55.37 (CH,C-5),
18.20 (CH2, C-6), 34.20 (CH2, C-7), 40.84 (C, C-8), 50.33
(CH, C-9), 37.08 (C, C-10), 20.94 (CH2, C-11), 25.10
(CH2, C-12), 38.04 (CH, C-13), 42.9 (C, C-14), 27.43 (CH2, C-15), 35.57 (CH2, C-16), 42.82 (C, C-17), 48.28
(C, C-18), 48.00 (C,C-19), 150.94 (C, C-20), 29.70 (CH2,
C-21), 40.00 (CH2, C-22), 27.97 (CH3, C-23), 16.57 (CH3, C-24), 16.17 (CH3, C-25), 15.97 (CH3, C-26), 14.51 (CH3,
C-27), 18.00 (CH3, C-28), 109.36 (CH2, C-29), 19.28
(CH3, C-30), 173.72 (C, C-1’), 34.86 (CH2, C-2’), 25.17 (CH2, C-3’), 29.26 (CH2, C-4’), 29.27 (CH2, C-5’), 29.38 (CH2, C-6’), 29.59 (CH2, C-7’), 14.13 (CH2, C-8’), 29.82 (CH2, C-9’), 29.70 (CH2, C-10’), 29.70 (CH2, C-11’),
29.70 (CH2, C-12’), 29.70 (CH2, C-13’), 29.59 (CH2,
C-14’), 29.47 (CH2, C-15’), 31.94 (CH2, C-16’), 22.70
(CH2, C-17’), 14.13 (CH3, C-18’) .
CHN analysis: Calcd for C48H84O2: C, 83.17; H, 12.21%.
Found: C, 82.97; H, 13.41%.
Lupeol palmitate (3): 664 g.mol-1, [0.90 mmol (90%
yield)], (mp 52.0 - 56.0 ºC).
IR (KBr, cm-1): 3071, 2915, 2850 (CH), 1726 (C=O), 1641,
1171 (CO-O-C), 881.
1H NMR (CDCl
3, 200 MHz) δ:4.57 (s, H-29a), 4.68(s,
H-29b), 4.48 ( dd, J= 6.0; 12.0 Hz, H-3), 1.68, 1.03, 0.94,
0.88, 0.85, 0.84, 0.79 ( 21H, 7 s, 7 CH3).
13C NMR (CDCl
3, 50 MHz) δ: 38.39 (CH2, C-1), 23.76
(CH2, C-2), 80.63 (C, C-3), 37.85 (C, C-4), 55.39 (CH,
C-5), 18.22 (CH2, C-6), 34.22 (CH2, C-7), 40.87 (C, C-8),
50.35 (CH, C-9), 37.10 (C, C-10), 20.96 (CH2, C-11), 25.11 (CH2, C-12), 38.01 (CH, C-13), 42.84 (C, C-14),
27.45 (CH2, C-15), 35.59 (CH2, C-16), 43.01 (C ,C-17) , 48.30 (C, CH-18), 48.32 (C, C-19) , 150.98 (C, C-20), 29.84 (CH2, C-21), 40.02 (CH2, C-22), 27.99 (CH3, C-23), 16.59 (CH3, C-24), 16.19 (CH3, C-25), 15.99 (CH3, C-26),
14.54 (CH3, C-27), 18.02 (CH3, C-28), 109.38 (CH2, C-29), 19.30 (CH3, C-30), 173.74 (C, C-1’), 34.88 (CH2,
C-2’), 25.01 (CH2, C-3’), 29.39 (CH2, C-4’), 29.49 (CH2, C-5’), 29.70 (CH2, C-6’), 29.61 (CH2, C-7’), 29.61 (CH2,
C-8’), 29.61 (CH2, C-9’), 29.61 (CH2, C-10’), 29.61 (CH2, C-11’), 29.61 (CH2, C-12’), 29.29 (CH2, C-13’), 31.95
(CH2, C-14’), 22.72 (CH2, C-15’), 14.15 (CH3, C-16’).
CHN analysis: Calcd for C46H80O2: C, 83.07; H, 12.12%.
Found: C, 83.25; H, 12.64%.
Lupeol miristate (4): 636 g mol-1, [0.95 mmol (95%
yield)], (mp 84.5 - 86.8 °C).
IR (KBr, cm-1): 2953, 2850(CH), 1728 (C=O),
1171(CO-O-C), 881.
1H NMR (CDCl
3, 200 MHz) δ:4.57 (s, H-29a), 4.68 (s,
H-29b), 4.48 ( dd, J= 6.0; 12.0 Hz, H-3), 1.68, 1.03, 0.94,
0.91, 0.88, 0.84, 0.79 ( 21H, 7 s, 7 CH3).
13C NMR (CDCl
3, 50 MHz) δ: 38.36 (CH2, C-1), 23.74
(CH2, C-2), 80.61 (C, C-3), 37.83 (C, C-4), 55.37(CH,
C-5), 18.20 (CH2, C-6), 34.20 (CH2, C-7), 40.84 (C, C-8),
50.32 (CH, C-9), 37.08 (C, C-10), 20.93 (CH2, C-11), 25.08 (CH2, C-12), 38.03 (CH, C-13), 42.82 (C, C-14),
27.43 (CH2, C-15), 35.56 (CH2, C-16), 43.00 (C, C-17), 48.28 (C, C-18), 48.01 (C, C-19), 150.98 (C, C-20), 29.83
(CH2, C-21), 40.00 (CH2, C-22), 27.97 (CH3, C-23), 16.58 (CH3, C-24), 16.17 (CH3, C-25), 15.97 (CH3, C-26), 14.52 (CH3, C-27), 18.00 (CH3, C-28), 109.36 (CH2, C-29),
19.28 (CH3, C-30), 173.74 (C, C-1’), 34.87 (CH2, C-2’),
25.18 (CH2, C-3’), 29.48 (CH2, C-4’), 29.37 (CH2, C-5’), 29.65 (CH2, C-6’), 29.60 (CH2, C-7’), 29.60 (CH2, C-8’),
29.60 (CH2, C-9’), 29.60 (CH2, C-10’), 29.27 (CH2, C-11’), 31.93 (CH2, C-12’), 22.70 (CH2, C-13’), 14.14
(CH2, C-14’).
CHN analysis: Calcd for C44H76O2: C, 82.95; H, 12.02%.
Found: C, 83.13; H, 12.49%.
Lupeol laurate (5): 608 g mol-1, [0.89 mmol (89% yield)],
(mp 93.4 - 95.1 °C).
IR (ATR, cm-1): 2923, 2853, 1727(C=O), 1176(CO-O-C),
1H NMR (CDCl
3, 200 MHz) δ:4.57 (s, H-29a), 4.68 (s,
H-29b), 4.47 ( m, H-3), 1.68, 1.02, 0.94, 0.88, 0.85,b0.84, 0.79 (21H, 7 s, 7 CH3).
13C NMR (CDCl
3, 50 MHz) δ: 38.37 (CH2, C-1), 23.75
(CH2, C-2), 80.60 (C, C-3), 37.83 (C, C-4), 55.37 (CH,
C-5), 18.20 (CH2, C-6), 34.21 (CH2, C-7), 40.84 (C, C-8),
50.33 (CH, C-9), 37.08 (C, C-10), 20.94 (CH2, C-11), 25.17 (CH2, C-12), 38.04 (CH, C-13), 42.82 (C, C-14),
27.43 (CH2, C-15), 35.57 (CH2, C-16), 43.00 (C, C-17), 48.28 (CH, C-18), 48.01 (C, C-19), 150.94 (C, C-20), 29.82 (CH2, C-21), 40.00 (CH2, C-22), 27.97 (CH3, C-23), 16.57 (CH3, C-24), 16.17 (CH3, C-25), 15.97 (CH3, C-26),
14.52 (CH3, C-27), 18.00 (CH3, C-28), 109.37 (CH2, C-29), 19.28 (CH3, C-30), 173.71 (C, C-1’), 34.86 (CH2,
C-2’), 25.17 (CH2, C-3’), 31.91 (CH2, C-4’), 22.69 (CH2, C-5’), 29.44 (CH2, C-6’), 29.59 (CH2, C-7’), 27.42 (CH2,
C-8’), 29.26 (CH2, C-9’), 31.90 (CH2, C-10’), 29.81 (CH2, C-11’), 14.12 (CH2, C-12’).
CHN analysis: Calcd for C42H72O2: C, 82.83; H, 11.92%.
Found: C, 83.07; H, 13.09%.
Lupeol caprate (6): 580 g mol-1, [0.92 mmol (92% yield)],
(mp 92.5 - 93.8 °C).
IR (ATR, cm-1): 2928, 2851, 1728(C=O), 1175(CO-O-C),
881.
1H NMR (CDCl
3, 200 MHz) δ:4.57 (s, H-29a), 4.68(s,
H-29b), 4.47 ( m, H-3), 1.68, 1.02, 0.94, 0.88, 0.85, 0.84, 0.79 ( 21H, 7 s, 7 CH3).
13C NMR (CDCl
3, 50 MHz) δ: 38.36 (CH2, C-1), 23.75
(CH2, C-2), 80.60 (C, C-3), 37.83 (C, C-4), 55.37 (CH,
C-5), 18.20 (CH2, C-6), 34.20 (CH2, C-7), 40.84 (C, C-8), 50.33 (CH, C-9), 37.08 (C, C-10), 20.94 (CH2, C-11),
25.17 (CH2, C-12), 38.04 (CH, C-13), 42.82 (C, C-14), 27.43 (CH2, C-15), 35.57 (CH2, C-16), 43.00 (C, C-17),
48.28 (CH, C-18), 48.00 (C, C-19), 150.94 (C, C-20), 29.82 (CH2, C-21), 40.00 (CH2, C-22), 27.97 (CH3, C-23),
16.57 (CH3, C-24), 16.17 (CH3, C-25), 15.97 (CH3, C-26), 14.52 (CH3, C-27), 18.00 (CH3, C-28), 109.36 (CH2,
C-29), 19.28 (CH3, C-30), 173.70 (C, C-1’), 34.87(CH2, C-2’), 25.17 (CH2, C-3’), 29.26 (CH2, C-4’), 29.19 (CH2,
C-5’), 29.43 (CH2, C-6’), 29.43 (CH2, C-7’), 31.86 (CH2, C-8’), 29.82 (CH2, C-9’), 14.11 (CH2, C-10’).
CHN analysis: Calcd for C40H68O2: C, 82.69; H, 11.80%.
Found: C, 82.77; H, 13.03%.
Lupeol caprilate (7): 552 g mol-1, [0.96 mmol (96%
yield)], (mp 145.0-145.7 °C).
IR (KBr, cm-1): 1727 (C=O); 1179 (CO-O-C), 2927, 2852
(CH).
1H NMR (CDCl
3, 200 MHz) δ:4.57 (s, H-29a), 4.68(s,
H-29b), 4.48 (dd, J= 6.0; 8.0 Hz, H-3), 1.68, 1.03, 0.94,
0.87, 0.85, 0.84, 0.79 (s, 21H, 7 s, 7 CH3). 13C NMR (CDCl
3, 50 MHz) δ: 38.36 (CH2, C-1), 23.73
(CH2, C-2), 80.60 (C, C-3), 38.02 (C, C-4), 55.36 (CH,
C-5), 18.19 (CH2, C-6), 34.20 (CH2, C-7), 40.83 (C, C-8), 50.32 (CH, C-9), 37.07 (C, C-10), 20.93 (CH2, C-11),
25.16 (CH2, C-12), 38.02 (CH, C-13), 42.82 (C, C-14), 27.42 (CH2, C-15), 35.56 (CH2, C-16), 42.98 (C, C-17),
48.27 (CH, C-18), 48.00 (C, C-19), 150.95 (C, C-20), 29.81 (CH2, C-21), 40.83 (CH2, C-22), 27.95 (CH3, C-23),
16.56 (CH3, C-24), 16.16 (CH3, C-25), 15.96 (CH3, C-26), 14.51 (CH3, C-27), 18.19 (CH3, C-28), 109.35 (CH2,
C-29), 19.27 (CH3, C-30), 173.70 (C, C-1’), 34.85 (CH2, C-2’), 25.16 (CH2, C-3’), 29.13 (CH2, C-4’), 29.13 (CH2,
C-5’), 31.67 (CH2, C-6’), 22.59 (CH2, C-7’), 14.06 (CH2, C-8’).
CHN analysis: Calcd for C38H64O2: C, 82.55; H, 11.67%.
Found: C, 82.68; H, 12.96%.
Lupeol caproate (8): 524 g mol-1,[0.87 mmol (87% yield)],
(mp 156.4 - 159.7 °C).
IR (KBr, cm-1): 2936, 2858, 1727(C=O), 1180(CO-O-C),
877.
1H NMR (CDCl
3, 200 MHz) δ:4.58 (d, J=2.0Hz, H-29a),
4.68 (d, J=2.0Hz, H-29b), 4.47 ( dd, J= 4.0; 6.0 Hz, H-3),
1.68, 1.03, 0.94, 0.85, 0.84, 0.83, 0.79 (21H, 7 s, 7 CH3) 13C NMR (CDCl
3, 50 MHz) δ: 38.37 (CH2, C-1), 23.74
(CH2, C-2), 80.62 (C, C-3), 37.83 (C, C-4), 55.37 (CH,
C-5), 18.20 (CH2, C-6), 34.21 (CH2, C-7), 40.85 (C, C-8), 50.33 (CH, C-9), 37.08 (C, C-10), 20.94 (CH2, C-11),
25.09 (CH2, C-12), 38.04 (CH, C-13), 42.83 (C, C-14), 27.43 (CH2, C-15), 35.57 (CH2, C-16), 42.99 (C, C-17),
48.28 (CH, C-18), 48.00 (C, C-19), 150.95 (C, C-20), 29.83 (CH2, C-21), 39.99 (CH2, C-22), 27.96 (CH3, C-23),
16.56 (CH3, C-24), 16.17 (CH3, C-25), 15.97 (CH3, C-26), 14.52 (CH3, C-27), 18.00 (CH3, C-28), 109.36 (CH2,
C-29), 19.28 (CH3, C-30), 173.71 (C, C-1’), 34.81 (CH2, C-2’), 24.83 (CH2, C-3’), 31.33 (CH2, C-4’), 22.32 (CH2,
C-5’), 13.92 (CH3, C-6’).
CHN analysis: Calcd for C36H60O2: C, 82.38; H, 11.52%.
Found: C, 82.66; H, 12.72%.
Lupeol 3’,4’-dimethoxybenzoate (9): 590 g.mol-1,[0.90
mmol (90% yield)],(mp 243.6 - 245.3 °C).
IR (KBr, cm-1): 2944, 2857, 1704, 1267, 1247, 968, 761.
1H NMR (CDCl
3, 200 MHz) δ: 7.70 (d, J=10.0 Hz, H-6´),
7.56 (s, H-2´), 6.90 (d, J=8.0Hz, H-5´) 4.69 (t, H-29a,b),
4.58 (s, H-3), 3.93 (s, H-9´, H-10´), 1.68, 1.05, 0.99, 0.97,
0.92, 0.90, 0.80 ( 21H, 7 s, 7 CH3).
13C NMR (CDCl
3, 50 MHz) δ: 38.40 (CH2,C-1),
(C, C-8), 50.36 (CH, C-9), 37.14 (C, C-10), 20.98 (CH2,
C-11), 23.83 (CH2, C-12), 38.06 (CH, C-13), 42.86 (C, C-14), 27.45 (CH2, C-15), 35.58 (CH2, C-16), 43.00 (C,
C-17), 48.29 (CH, C-18), 48.02 (C, C-19), 150.95 (C, C-20), 29.84 (CH2, C-21), 40.01 (CH2, C-22), 28.14 (CH3,
C-23), 16.01 (CH3, C-24), 16.81 (CH3, C-25), 16.20 (CH3, C-26), 14.56 (CH3, C-27), 18.02 (CH3, C-28), 109.38
(CH2, C-29), 19.30 (CH3, C-30), 166.10 (C, C-1’), 152.79
(C, C-2’), 112.03 (CH, C-3’), 148.59 (C, C-4’), 150.95 (C, C-5’), 110.21 (CH, C-6’), 123.35 (CH, C-7’), 55.94 (CH3, C-8’), 55.94 (CH3, C-9’).
CHN analysis: Calcd for C39H58O4: C, 79.28; H, 9.89%.
Found: C, 79.33; H, 10.56%.
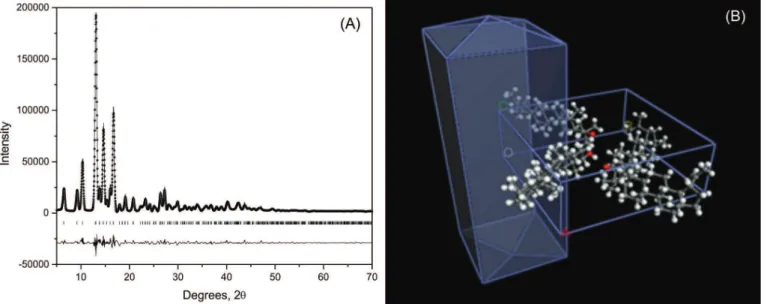
XRD analysis of compound 1 revealed its needle
shape and established a structure in which the carbon atoms distribution (Figure 2) is in accordance with the
13C NMR data.
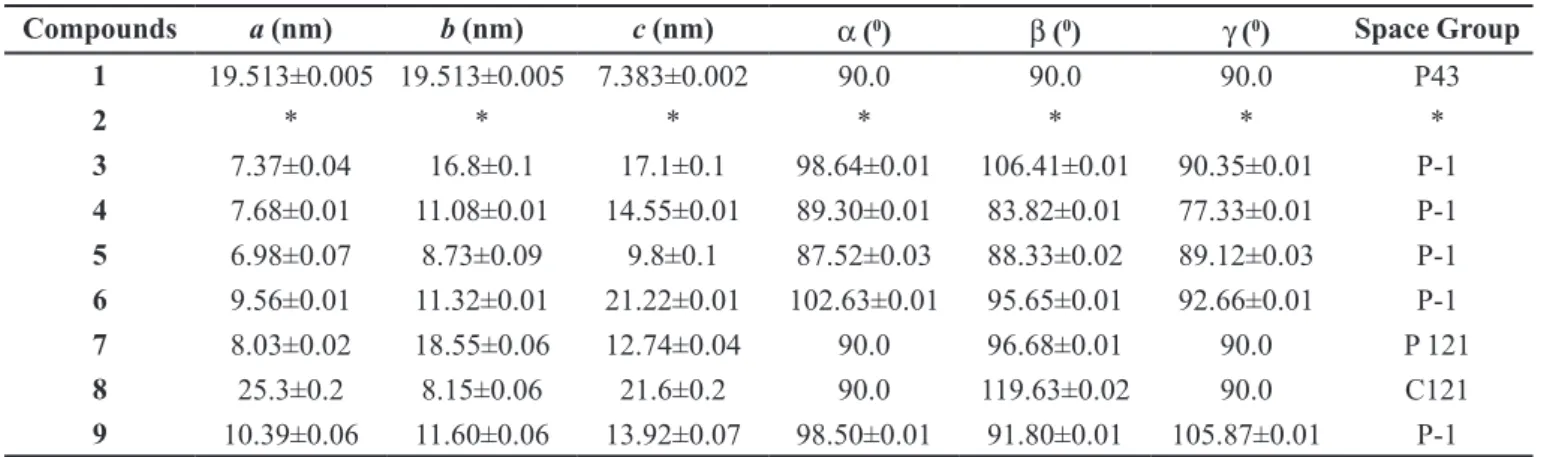
For the XRD experiments, the material was homogenously spread over the sample holder under spinning to prevent preferred orientation and minimize rugosity effects over the exposed surface. The small amount submitted to the XRD, few milligrams, was composed essentially of polycrystalline material. Single crystals were not identiied or isolated from the synthetic material. So, detailed crystallographic data were provided only for the isolated lupeol. For lupeol (1), the angles are
90.0 due to the special positions on tetragonal P43 space group symmetric restrictions. Other details of reinements and X-ray difraction experimental data are summarized in Table I. Due to the small amount of esters (2 to 9),
all ittings were obtained at P-1 space group that safely
allowed us to index all peaks. After extracting and itting, all peaks in space group P-1 were searched for more symmetric space group based on the Bragg systematic absences. More symmetric space groups were achieved for compounds 1, 7 and 8. The remaining ones have not shown
any symmetric description based on the systematic Bragg absences. The powder XRD data of lupeol esters 2 to 9
(Table I) were consistent with the 13C NMR data of each
one indicating the tendency of the compounds to be in the crystalline state. The XRD experiment was considered as an excellent tool to determine the structure of lupeol and
its esters in solid state.
Cell proliferation assays
All the compounds were tested for proliferation of human cancer cells. Doxorubicin (anthracycline) used as positive control is a chemotherapy drug that decreases or stops the growth of cancer cells. The activity of doxorubicin involves blocking the enzyme called topo isomerase 2 that cancer cells need to replicate and grow. Lupeol was inactive (GI50>250 µg/mL) in the experimental condition while lupeol esters 2-4 and 7-9
showed a cytostatic efect on colorectal adenocarcinoma (HT-29) and chronic myelogenous leukemia (K-562) cell lines (Table II).
The introduction of a long alkyl side chain (2) in
lupeol resulted in a cytostatic effect on the colorectal adenocarcinoma (HT29) cell line (GI50 = 97.81 µg/mL).
This efect increased by reducing the length of the alkyl chain, from C16 (2) to C12 (4), resulting in the best
effect (GI50 = 1.74 µg/mL). However, the continuous
FIGURE 2 - Powder X-ray difraction (XRD) data: (A) lupeol difratogram; (B) lupeol chemical structure, found in solid with a
reduction in the chain length (from C10 to C4, compounds
5-8) afforded inactive compounds (GI50>250 µg/mL).
Moreover, lupeol palmitate (3) showed a selective
growth inhibition effect on erythromyeloblastoid leukemia (K-562) cells in a concentration-dependent way (Figure S2). Previous studies have shown that the palmitic acid is active against leukemic cells (Harada
et al., 2002). Probably, the activity observed for lupeol
palmitate is due to the fatty moiety. Thus, compound 3
could be considered as a prototype for the development of new anticancer drugs to be used in leukemia treatment. For the aryl lupeol ester (9) it was seen a quite similar
cytostatic efect (GI50=0.95 µg/mL) to that observed for
ester 4. On the other hand, the side chain length seemed
not to be as inluent against chronic myelogenous leukemia (K-562) cells as for antiproliferative activity against HT-29. In this regard, only compounds 5 (with ten carbon in
side chain) and 6 (with eight carbons in side chain) were
not able to inhibit K-562 cell proliferation up to 250 µg/ mL (Figure S2, Table II). The esters 4 and 9 showed a
selective cytostatic efect with low GI50 values (Figure S2), therefore, these compounds represent a promising prototype for the development of new anticancer drugs.
For the aryl lupeol ester (9) it was seen a quite
similar cytostatic efect (GI50 = 0.95 µg/mL) than observed
for ester 4.
TABLE I - Lupeol (1) and its esters (2 to 9) lattice parameters obtained by Rietveld itting of the powder X-ray difraction
Compounds a (nm) b (nm) c (nm) α (0) β (0) γ (0) Space Group
1 19.513±0.005 19.513±0.005 7.383±0.002 90.0 90.0 90.0 P43
2 * * * * * * *
3 7.37±0.04 16.8±0.1 17.1±0.1 98.64±0.01 106.41±0.01 90.35±0.01 P-1
4 7.68±0.01 11.08±0.01 14.55±0.01 89.30±0.01 83.82±0.01 77.33±0.01 P-1
5 6.98±0.07 8.73±0.09 9.8±0.1 87.52±0.03 88.33±0.02 89.12±0.03 P-1
6 9.56±0.01 11.32±0.01 21.22±0.01 102.63±0.01 95.65±0.01 92.66±0.01 P-1
7 8.03±0.02 18.55±0.06 12.74±0.04 90.0 96.68±0.01 90.0 P 121
8 25.3±0.2 8.15±0.06 21.6±0.2 90.0 119.63±0.02 90.0 C121
9 10.39±0.06 11.60±0.06 13.92±0.07 98.50±0.01 91.80±0.01 105.87±0.01 P-1
TABLE II - Concentration (µg/mL) of lupeol (1) and its esters (2 - 9) necessary to inhibit 50% cell growth (GI50)
Compound tested
Cell lines
U251 NCI-ADR/
RES 786-0 NCI-H460 PC-3 OVCAR-03 HT29 K562
Doxo <0.025 25 0.038 <0.025 0.025 0.23 0.026 >25
1 * * * * * * * *
2 * * * * * * 97.81 0.35
3 * * * 250 * * 35.94 <0.25
4 * * * * * * 1.74 0.41
5 * * * * * * * *
6 * * * * * * * *
7 * * * * * * * <0.25
8 * * * * * * 250 <0.25
9 * * * * * * 0.95 <0.25
Key: * = GI50>250 µg/mL; Doxo, doxorubicine (positive control); 3β-Lup-20(29)-en-3-ol (lupeol) (1); lupeol stearate (2);
lupeol palmitate (3); lupeol miristate (4); lupeol laurate (5); lupeol caprate (6); lupeol caprilate (7); lupeol caproate (8); lupeol
CONCLUSION
The esters 2 to 9 were obtained using lupeol,an
adequate carboxylic acid and DIC/DMAP reagents, with yields ranging from 86 to 96%. The esters 5 to 9 were
new compounds. The XDR method was an excellent tool to determine the structure of lupeol and its esters in solid
state. Lupeol esters 3, 4 and 9 showed a selective cytostatic
effect with low GI50 values, representing a promising
prototype for the development of new anticancer drugs.
EXPERIMENTAL SECTION
General experimental procedures
Melting points (mp) (uncorrected) were determined using a Mettler FP 80 HT apparatus. 13C NMR spectra were
obtained on a Bruker Avance DRX 400 or on Bruker DPX
200 spectrometers. The sample was dissolved in CDCl3
and TMS was used as internal standard (δC = 0). IR spectra
were recorded on a FITR–Perkin-Elmer, Spectrum One SN 74759 spectrophotometer. Powder X-ray difraction (XRD) data were collected in an XRD-7000 difractometer (Shimadzu, Japan) under 40 kV, 30 mA, using Cu Kα (λ = 1.54056 Å) equipped with a polycapillary focusing optics under parallel geometry coupled with a graphite monochromator, scanned over an angular range of 4−70° (2θ) with a step size of 0.01° (2θ) and a time constant of 5 s.step−1. The sample holder was submitted to a spinning
of 30 cycles per minute to minimize rugosity efects and to reduce any eventual preferred orientation. The lattice parameters were extracted and itted by Rietveld itting analysis. CHN analyses were performed in a Perkin Elmer, Series II, CHNS/O Analyzer. Classical chromatographic column (CC) was carried out using silica gel 60 (Merck, 70-230 Mesh). TLC was obtained using pre-coated silica gel plates, and the detection was visualized by spraying the plates with solution (1:1) of vanillin (ethanol 1 % solution w/v) in perchloric acid (3% aqueous solution v/v), in accordance with Wagner and Bladt (1996).
Plant material
Maytenus salicifolia Reissek (Celastraceae) was
collected at ‘Serra de Ouro Branco’, a mountain located in the Ouro Branco City region, Minas Gerais (MG) state, Brazil. The plant was identiied by Dr. Rita Maria Carvalho-Okano, Botanist of the Universidade Federal de Viçosa, MG, Brazil. A voucher specimen of M. salicifolia was
deposited (Nº. OUPR-18094) at the Herbarium José Badini of the Universidade Federal de Ouro Preto, MG, Brazil.
Isolation of lupeol and synthesis of esters
The isolation of lupeol was reported by Magalhães
and coworkers (2011). For the esters synthesis, the following sequence was carried out for the reactions: to 1.0
mmol of lupeol (1), x mmol of carboxylic acid and y mmol
of 4-(dimethylamino)pyridine (DMAP) in 7.0 mL of dry dichloromethane were added (Table I). After cooling down to 0 °C and under constant magnetic stirring, z mmol of N,N‘-diisopropylcarbodiimide (DIC) was carefully added.
Then, the reaction mixture was maintained under magnetic stirring, at room temperature, for 2 to 48 hours depending on the carboxylic acid used as reagent. The reaction time was monitored by TLC using CHCl3-MeOH (9.5:0.5) as mobile phase. The reaction conditions of carboxylic acid with lupeol [1, (1.0 mmol)] and DIC/DMAP to obtain the lupeol esters 2 to 9 (Figure 1) are presented in Table SI.
At the end of the reaction, the dichloromethane was recovered in a rotator evaporator and the residual material obtained from each esteriication reaction was puriied by chromatographic column eluted with CHCl3. The lupeol
esters 2 to 4 (Figure 1) were obtained as a white waxy
material while lupeol esters 5 to 9 were obtained as a white amorphous solid.
Characterization of compounds
The structure of lupeol and its synthesized esters were initially characterized by IR, NMR (1H, 13C) and
CHN data. The spectral results were carefully compared with data available in the literature (Mahato, Kundu, 1994). Then, the structure of each compound was itted through powder XRD. Thus, compound 1 (or ester 2 to 9) was reduced to a very fine powder and deposited
small amount of material (lupeol esters), all ittings were obtained at P-1 space group, which allowed us to index all peaks safely. To search for more symmetric Space Group occurrences more natural extracted material would be necessary to increase low intensity peaks that may help search for more symmetry in all diffractograms. After extracting and itting all peaks in space group P-1, a search was performed for more symmetric space groups based on the Bragg systematic absences. Details of reinements and experimental data of X-ray difraction are summarized in Table I.
Antiproliferative activity
Human cell lines
Eight human tumor cell lines were used: U-251 (glioma), NCI-ADR/RES (ovarian expressing the resistance phenotype for adryamycin), 786-0 (kidney), NCI-H460 (lung, non-small cells), PC-3 (prostate), OVCAR-03 (ovarian), HT-29 (colon), and K-562 (erythromyeloblastoid leukemia). The eight human tumor cell lines were provided by the Frederick Cancer Research & Development Center, National Cancer Institute, Frederick, MA, USA. The cells were grown in RPMI 1640 Medium (GIBCO BRL) supplemented with 5% fetal bovine serum (FBS) (GIBCO BRL) and penicillin/ streptomycin mixture (1000 U/mL: 1000 µg/mL, 1.0 mL/L RPMI) at 37 oC in a 5% CO
2 atmosphere.
Sample preparation
Aliquots (5.0 mg) of lupeol and its esters 2 to 9 were
initially diluted in DMSO (50 µL) followed by the addition of 950 µL of RPMI 1640/FBS 5% (working solution). The solutions were then diluted in RPMI 1640/FBS 5% in order to obtain the inal concentrations. DMSO inal concentrations (≤ 0.25%) in culture medium did not afect the cell viability.
Antiproliferative assay
Cells in 96-well microplates (100 µL cells/well, inoculation density from 3 to 7 x 104 cell/mL) were
exposed for 48 h to crescent concentrations (0.25, 2.5, 25.0, and 250.0 µg/mL, in triplicate) of 1 and its esters 2 to 9 at 37 0C in a 5% CO
2 atmosphere. Doxorubicin
chloridrate (0.1mg/mg; Europharma) was used as positive control (0.025, 0.25, 2.5 and 25 µg/ml). Before (T0 plate) and after sample addition (T1 plates), cells were fixed with 50% trichloroacetic acid (50 µL/well). Cellular proliferation was determined by the spectrophotometric quantiication (540 nm) of cellular protein content using sulforhodamine B assay (Monks et al., 1991). Using
the concentration-response curve for each cell line, GI50
(concentration that inhibits cell growth by 50%) was determined through non-linear regression analysis using the software ORIGIN 8.0 (Origin Lab Corporation) (Dos
Santos et al., 2015; Da Silva et al., 2015).
ACKNOWLEDGEMENTS
The authors thank the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) and Fundação de Amparo à Pesquisa de Minas Gerais (FAPEMIG) for inancial support.
REFERENCES
Andrikopoulos NK, Kaliora AC, Assimopoulou AN, Papapeorgiou VP. Biological activity of some naturally occurring resins, gums and pigments against in vitro LDL oxidation. Phytother Res. 2003;17(5):501-507.
Chabner BA, Roberts Jr TG. Chemotherapy and the war on cancer. Nat Rev Cancer. 2005;5:65-72.
DallaVechia L, Gnoatto SCB, Gosmann G. Derivados oleananos e ursanos e sua importância na descoberta de novos fármacos com atividade antitumoral, anti-inlamatória e antioxidante. Quím Nova. 2009;32(5):1245-1252.
Da Silva DL, Terra BS, Lage MR, Ruiz ALTG, Da Silva CC, De Carvalho JE, et al. Xanthenones: Calixarenes-catalyzed syntheses, anticancer activity and QSAR studies. Org & Biomol Chem. 2015;13(11):3280-3287.
Dos Santos DS, Piovesan LA, D’oca CRM, Hack CRL, Treptow TGM, Rodrigues MO, et al. Antiproliferative activity of synthetic fatty acid amides from renewable resources. Bioorg
& Med Chem. 2015;23(2):340-347.
Ferlay J, Soerjomataram I, Ervik M, Forman D, Bray F, Dikshit R, et al. GLOBOCAN 2012: Estimated cancer incidence, mortality and prevalence worldwide in 2012. [cited 2016 July 08]. Available from: http://globocan.iarc.fr/Pages/fact_sheets_ cancer.aspx.
Fotie J, Bohle DS, Leimanis ML, Georges E, Rukunga G,
Gali-Muhtasib H, Hmadi R, Kareh M, Tohme R, Darwiche N. Cell death mechanisms of plant-derived anticancer drugs: beyond apoptosis. Apoptosis. 2015;20(12):1531-1562.
Gallo MBC, Sarachine MJ. Biological activities of lupeol.Int J Biomed Pharmaceut Sci. 2009;3:46-66.
Galmarini D, Galmarini CM, Galmarini FC. Cancer chemotherapy: a critical analysis of its 60 years of history. Crit Rev Oncol Hemat. 2012;84(2):181-199.
Geetha T, Varalakshmiu P, Latha RM. Efect of triterpenes from
Crataeva nurvala stem bark on lipid peroxidation in adjuvant
induced arthritis in rats. Pharmacol Res. 1998;37(3):191-195.
Gurovic MS, Castro MV, Richmond VJ, Faraoni MB, Maier MS, Murray AP. Tritepenoids with acethylcholinesterase inhibition from Chuquiraga erinacea D. Don. Supbsp. Erinacea (Asteraceae). Planta Med. 2010;76(6):607-610.
Harada H, Yamashita U, Kurihara H, Fukushi E, Kawabata J, Kamei Y. Antitumor activity of palmitic acid found as a selective cytotoxic substance in a marine red alga. Anticancer Res. 2002;22(5):2587-2590.
Lachance H, Wetzel S, Kumar K, Waldmann H. Charting, navigating, and populating natural product chemical space for drug discovery. J Med Chem. 2012;55(13):5989-6001.
Laszczyk MN. Pentacyclic triterpenes of the lupane, oleanane and ursane group as tools in cancer therapy. Planta Med. 2009;75(15):1549-1560.
Li W, Hao J, Xiao Y. Synthesis and in vitro antitumor activities of lupeol dicarboxylic acid monoester derivatives. Arch Pharmacol Res. 2013;36(12):1447-1453.
Magalhães CG, Ferrari FC, Guimarães DAS, Silva GDF, Duarte LP, Figueiredo RC, et al. Maytenus salicifolia: triterpenes
isolated from stems and antioxidant property of extracts from aerial parts. Braz J Pharmacogn. 2011;21(3):415-419.
Mahato SB, Kundu AP. 13C NMR Spectra of pentacyclic triterpenoids - a compilation and some salient features. Phytochemistry. 1994;37(6):1517-1575.
Monks A, Scudeiro D, Skehan P, Shoemaker S, Paull K, Vistica D, et al. Feasibility of a high-lux anticancer drug screen using a diverse panel of cultured human tumor cell lines. J Nat Can Inst. 1991;83(11):757-766.
Newman DJ, Cragg GM. Natural products as sources of new drugs from 1981 to 2014. J Nat Prod. 2016;79(3):629-661.
Núñez MJ, Reyes CP, Jiménez IA, Moujir L, Bazzocchi IL.
Lupane triterpenoids from Maytenus species. J Nat Prod.
2005;68(7):1018-1021.
Oishi-Tomiyasu R. Rapid Bravais-lattice determination algorithm for lattice parameters containing large observation errors. Acta Cryst A. 2012;68(Pt 5):525-535.
Reddy KP, Singh AB, Puri A, Srivastava AK, Narender T. Synthesis of novel triterpenoid (lupeol) derivatives and their in vivo antihyperglycemic and antidyslipidemic activity. Bioorg Med Chem Lett. 2009;19(15):4463-4466.
Saleem M, Maddodi N, Zaid MA, Khan N, Hafeez B, Asim M, et al. Lupeol inhibits growth of highly aggressive human metastatic melanoma cells in vitro and in vivo by inducing apoptosis. Clin Cancer Res. 2008;14(7):2119-2127.
Saleem M. Lupeol, a novel anti-inlammatory and anti-cancer dietary triterpene. Cancer Lett. 2009;285(2):109-115.
Salminen A, Lehtonen M, Suuronen T, Kaarniranta K, Huuskonen J. Terpenoids: natural inhibitors of NF-kB signaling with anti-inlammatory and anticancer potential. Cell Mol Life Sci. 2008;65(19):2979-2999.
Shahlaei M, Ghanadian SM, Ayatollahi AM, Mesaik MA, Abdalla OM, Afsharypour S, et al. Molecular modeling, structure activity relationship and immunomodulatory properties of some lupeol derivatives. Med Chem Res. 2013;22:1795-1803.
Siddique HR, Saleem M. Beneficial health effects of lupeol triterpene: a review of preclinical studies. Life Sci. 2011;88(7-8):285-293.
Sudhahar V, Kumar SA, Varalakshmi P. Role of lupeol and lupeol linoleate on lipemic-oxidative stress in experimental hypercholesterolemia. Life Sci. 2006a;78(12):1329-1335.
Sudhahar V, Kumar SA, Varalakshmi P. Efect of lupeol and lupeol linoleate on lipemic - hepatocellular aberrations in rats fed a high cholesterol diet. Mol Nutr Food Res. 2006b;50(12):1212-1219.
Sudhahar V, Kumar SA, Mythili Y, Varalakshmi P. Remedial efect of lupeol and its ester derivative on hypercholesterolemia-induced oxidative and inflammatory stresses. Nut Res 2007b;27(12):778-787.
Sudhahar V, Kumar SA, Varalakshmi P, Sujatha V. Protective efect of lupeol and lupeol linoleate in hypercholesterolemia associated renal damage. Mol Cell Biochem 2008;317(1-2):11-20.
Sudhahar V, Veena CK, Varalakshmi P. Antiurolithic efect of lupeol and lupeol linoleate in experimental hyperoxaluria. J Nat Prod 2008;71(9):1509-1512.
Sunitha S, Nagaraj M, Varalakshmi P. Hepatoprotective efect of lupeol and lupeol linoleate on tissue antioxidant defense system in cadmium-induced hepatotoxicity in rats. Fitoterapia. 2001;72(5):516-523.
Sultana NJ. Clinically useful anticancer, antitumor, and antiwrinkle agent, ursolic acid and related derivatives as medicinally important natural product. Enz Inhibit Med Chem. 2011;26(5):616-642.
Wagner H, Bladt S. Plant drug analysis: a thin layer chromatography atlas. Berlin: Springer; 1996. 384 p.
Received for publication on 08th February 2017