Revista
Brasileira
de
Hematologia
e
Hemoterapia
Brazilian
Journal
of
Hematology
and
Hemotherapy
w w w . r b h h . o r g
Case
report
Gaucher
disease
in
a
family
from
Maranhão
Samira
Shizuko
Parreão
Oi
a,
Dario
Itapary
Nicolau
b,
Sebastião
Kelson
Alves
dos
Santos
a,
Marcos
Antonio
Custódio
Neto
da
Silva
b,
Grac¸a
Maria
de
Castro
Viana
b,
Maria
do
Desterro
Soares
Brandão
Nascimento
b,∗ aUniversidadeEstadualdoMaranhão(UEMA),Caxias,MA,BrazilbUniversidadeFederaldoMaranhão(UFMA),SãoLuis,MA,Brazil
a
r
t
i
c
l
e
i
n
f
o
Articlehistory:
Received6November2013 Accepted24March2014 Availableonline19July2014
Keywords:
Gaucherdisease
Enzymereplacementtherapy Family
a
b
s
t
r
a
c
t
Background:Gaucherdiseaseisan inborn,autosomal recessiveerrorofthe metabolism whichbelongstothegroupoflysosomalstoragedisorders.
Objective:ThisworkreportsonthetreatmentofGaucherdiseaseinseveralmembersofthe samefamilyfromthecountrysideofMaranhão.
Methods:Thiswasanobservational,retrospectiveandprospective,descriptivecasestudy abouttheefficacyofenzymereplacementtherapy.
Results:Theresultsshowedthatwomenweremoreaffected(80%ofpatients)bythedisease, ageatdiagnosisrangedfrom24to33years,thepredominantethnicitywasmulatto(80%) andallcaseswereclassifiedastype1.Thediagnosisofthesepatientswasperformedby measuringthelevelsofglucocerebrosidaseandchitotriosidaseenzymesandconfirmedby genotyping.AllpatientssufferingfromGaucherdiseasehadlowglucocerebrosidaselevels. Beforereplacement therapy,hepatosplenomegalywasthemostcommonclinical mani-festation(100%)andosteopeniawasseenin80%ofthecases.Regardinghematological manifestations,anemiaandleukopeniawerefoundin40%ofpatientsatdiagnosis;however thehemoglobinandleukocytelevelswerenormalizedafterfouryearsoftherapy. Throm-bocytopenia,observedin20%ofcases,wasnormalizedafterthesecondyearoftreatment.
Conclusion: Inthesecases,despitegapsinthetreatmentasthefamilyresidesintherural region ofthestate, thepatients withGaucherdiseaseshowed satisfactory therapeutic responseovertime.
©2014Associac¸ãoBrasileiradeHematologia,HemoterapiaeTerapiaCelular.Published byElsevierEditoraLtda.Allrightsreserved.
Introduction
Gaucherdisease(GD)wasfirstdescribedin1882.Itisa reces-siveautosomallysosomalstoragedisorderwhichiscausedby
∗ Correspondingauthorat:NúcleodeImunologiaBásicaeAplicada,UniversidadeFederaldoMaranhão(UEMA),Av.dosPortugueses,1966,
CCBS,bloco3,sala3A,Bacanga,65080-805SãoLuís,MA,Brazil.
E-mailaddress:[email protected](M.doDesterroSoaresBrandãoNascimento).
glucocerebrosidasedeficiencythatinducesanaccumulation ofundigestedglycolipidsandleadstohistologicalchanges, especiallyevidentinorgansthatarerichinelementsofthe monocytic-phagocyteimmunesystem(spleen,liverandbone
http://dx.doi.org/10.1016/j.bjhh.2014.07.011
marrow).1,2Inmoreseverecases,thisdiseasecanaffectthe lungs,kidneysandcentralnervoussystem(CNS).3–5
GDisclassifiedintothreetypesaccordingtothepresence orabsenceofprimarydisease intheCNS:Type 1,the non-neuropathicformisthemostcommon,Type2,presentswith seriousimpairmentofCNSinchildhood,andType3,withmild impairmentoftheCNSinadolescenceorearlyadulthood.6
The most common type is also called the adult’s non-neuropathic chronicform(90–99% ofcases). Itisprevalent amongcasesofAshkenaziJews,affectingabout1:450ofthese individualsand1:40,000ofthegeneralpopulation.7,8
Diagnosis isby biochemical tests that can demonstrate glucocerebrosidasedeficiencywithenzymeactivityin leuko-cytesandculturedskinfibroblastsbelow10%ofthenormal level.Thesevaluesaredeterminedbylaboratorytestsorby skinbiopsy.4,9Abonemarrowbiopsyiswidelyusedto iden-tifyGauchercells,however,it isnotpathognomonicandit canoftenleadtowrongdiagnosessuchaschronicmyeloid leukemia, myeloblastic leukemia, Niemann-Pick disease, Hodgkin’sdiseaseandnon-Hodgkinnodularlymphoma.7
GD is transmitted by autosomal recessive inheritance definedbymutationsinthebeta-glucosidase(GBA)genethat isresponsibleforencodingtheglucocerebrosidaseenzyme. Thisgeneislocatedonthelongarmofchromosome1(q21 region).Thepresenceoftwomutantallelesconfirms diagno-sis.ThemostprevalentmutationsinthisdiseaseareN370S andL444P.10
The main clinical manifestations of GD include: hep-atosplenomegaly,hematologicaldisorders(anemia, thrombo-cytopenia and,more rarely, leukopenia), bone injuriesand CNSimpairment.11 Besides these symptoms, patients may alsobeaffectedbynon-specificsymptomssuchasinsomnia, headaches,chronicpain,paresthesia,depressionanddiffuse musculoskeletal pain. These patients do not have a good responsetoenzymereplacementtherapy(ERT).
Inaddition,psychiatricsymptoms,andmoresubtle,initial cognitiveandmotorchangesmayalsoappear.12,13
Thetreatment of mostsymptomatic patients ismainly supportivecarewithpainkillers.Thus,theBrazilianMinistry ofHealth introduced ERTfor GDin1990 throughthe Spe-cialDrugProgram,producingaprotocolofcorrecttherapeutic procedures.14,15
ERThassignificantlychangedtreatmentforGaucher dis-easewithreducedmorbidityandimprovedqualityoflifeand thusthisiscurrentlythetreatmentofchoice;itisfreethrough theBrazilianNationalHealthService.16
HereinwereporttheresponsetoERTofagroupofpatients withGaucherdiseaseinthesamefamilyfromSãoDomingos doMaranhão.SãoDomingosdoMaranhaoislocated380km fromthecapital,SãoLuís,andaccordingtothelatestcensus, thepopulationis33,506people.17,18Thisstudywasapproved inallrespectsbytheEvaluationCommitteeforResearchand Training(CAPPE)ofHEMOMAR.
Case
report
ThisstudyincludedfivepatientswiththediagnosisofGDofa familyof11individualsfromthemunicipalityofSão Domin-gosdoMaranhão.All were treatedbythe Hematologyand
1 I
II
1
N/N370S N/? N370S/? N/N
(?) means unidentified mutation (N) Normal allele
3
2 4 5 6 7 8 9
2
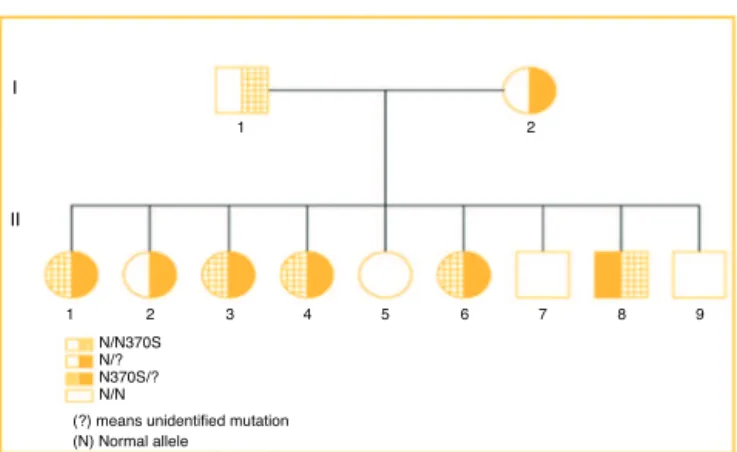
Figure1–Familyheredogramshowingsegregationofthe
twomutations.
HemotherapyCenterofMaranhão(HEMOMAR)locatedinthe statecapital.
Theinstrumentusedfordatacollectionincludedtwoforms tocollectclinical,laboratory,radiologicalandsocioeconomic data, besides the medical records from 2003to 2010. Data on gender,race,age atdiagnosis,occupation,classification accordingtothediseasetype,weight,height, glucocerebrosi-daseand chitotriosidaseenzymelevels andtypeofgenetic mutationwereobtainedfrommedicalrecords.
The family heredogram below shows three individuals heterozygous forGD(I1, I2and II2),five compounded het-erozygous forGD(II1,II3,II4,II6andII8) andthreenormal individuals(II5,II7andII9)(Fig.1).
All patients had diagnostic confirmationofGD by mea-surementofglucocerebrosidaseenzymeactivity.Theaffected family members (heterozygotes) are:II1, II3,II4, II6,II8, all featuring the same genotype (N370S/?). Patient II2 is the onlymemberofthesecondheterozygousgenerationforthe unidentifiedallele(?),whosegenotypeisnormal/?Theother membersofthesecondgeneration:II5,II7andII9arenormal, withnosignificantchangesintheirenzymemeasurements. ParentsI1andI2areheterozygousforN370Sand? respec-tively.AllpatientsaffectedbyGDshowedveryhighlevelsof chitotriosidase,exceptforPatientII4(Table1).
Thereispredominanceoffemales(80%).Table2showsthat allpatientswerediagnosedinadulthoodwithagesbetween24 and33yearsandameanof28.4yearsandarepredominantly mulattowithonlyoneCaucasianpatient.Mostpatientsare agriculturallaborers(80%).Allpatientsaretype1orcompose acharacteristicnon-neuropathicclinicalpicture.
Dataonthedeterminationofglucocerebrosidaseand chi-totriosidaselevelswerecollectedfrommedicalrecords.Fig.2
showsthatonlyPatient#3hadanormalchitotriosidaselevel, whiletheothershadhighlevelsofthisenzymebefore start-ingERT.Afteraboutsevenyears(incaseofPatients#1–4)and threeyears(Patient#5)ofERT,allpatientshadchitotriosidase levelswithinthenormalrange.
Thepresenceofanemia, leukopenia, thrombocytopenia, splenomegaly,hepatomegalyandskeletalchangeswere eval-uatedpriortoandafterstartingERTbetween2003and2010.
Table1–Glucocerebrosidaseandchitotriosidaselevelsandthegenotypeofallfamilymembers.
Familymember Glucocerebrosidase(VN: 10–45)(nmol/h/mgprotein)
Chitotriosidase(VN: 8.85–132)(nmol/mL/h)
Interpretation Genotype
I1 7.9 12.8 HeterozygousforN370S Normal/N370Sa
I2 5.26 5.6 Heterozygousfor? Normal/?a
II1 1.4 3150 Compoundheterozygous N370S/?
II2 4.0 2 Heterozygousfor? Normal/?
II3 2.1 7462 Compoundheterozygous N370S/?
II4 0.39 48 Compoundheterozygous N370S/?
II5 10.1 – Normal Normal/normala
II6 1.2 8694 Compoundheterozygous N370S/?
II7 10.2 12.9 Normal Normal/normala
II8 0.5 1897 Compoundheterozygous N370S/?
II9 6.88 – Normal Normal/normala
a Patientsinwhomthegenotypewasdeducedaccordingtotheenzymaticdosage. ?meansunidentifiedmutation.
Thenormalvalueusedbythelaboratorythatanalyzedtheactivityofglucocerebrosidaseis>3.2nmol/mg/h.
Table2–Sociodemographiccharacteristicsandclinical classificationofpatientswithGaucherdisease.
n %
Gender
Male 1 20
Female 4 80
Ageatdiagnosis
20–25years 1 60
26–30years 3 20
>30yearsand 1 20
Race
Caucasian 1 20
Mulatto 4 80
Profession
Farmer 4 80
Housewife 1 20
normalizationofhemoglobinlevelsinallpatientswiththese levelsremainingnormaluntil2010,exceptforPatient#4,who showedadecreasedhemoglobinlevelin2008.
Atdiagnosis,40%ofpatientswereleukopenicbutafterfour yearsofERT,nopatienthadleukopenia.In2008,Patients#1 and#4presentedwithleukopenia.
Table3–BoneimpairmentinpatientswithGaucher
diseasebeforeandafterenzymereplacementtherapy
(ERT).
n %
ImagingchangesafterERT
Absent 1 20
Osteopenia 4 80
Avascularnecrosis 1 20
DeformitiesinErlenmeyerflask 1 20
Lyticlesions 1 20
Fractures 1 20
ImagechangesbeforeERT
Absent 2 60
Avascularnecrosis 1 20
DeformitiesinErlenmeyerflask 2 60
9,000
8,000
7,000
6,000
5,000
4,000
3,000
2,000
1,000
0
Patient 1
Patient 1
3,150 Chitotriosidase before treatment
Chitotriosidase -2010 21 7.8
7,462 48 8,694 1,897
0.2 82 24.6
Patient 2
Patient 3
Patient 4
Patient 5
Levels of chitotriosidase (nmol/h/mL)
Patient 2 Patient 3 Patient 4 Patient 5 NV: 8.85-132
Figure2–Distributionofchitotriosidaselevelsbefore enzymereplacementtreatmentandin2010.
Atdiagnosis,20%ofpatientshadthrombocytopeniawhich wasresolved afterthe secondyearofERTwithallpatients havingnormalplateletcounts.ItisnoteworthythatPatient #1hadnothrombocytopeniabeforeERThowevertheplatelet countdroppedin2006.
At diagnosis, there was bone impairment in 80% of patients. Themostcommon osseousimpairment priorthe ERTwasosteopenia,presentin80%ofpatients(Table3).It seemsthatboneimpairmentisstillpresentduringERT,with 20%ofcaseshavingErlenmeyerflaskdeformityand20% hav-ing avascular necrosis. Some patients had more than one lesionbeforetheERTbuttherewerenoimagingchangesin 60%ofcasesafterERT(Table3).
Visceromegaliesandhepatosplenomegalywerepresentin allofthepatientsbeforeERThoweverattheendofthisstudy only20%ofpatientshadsplenomegaly.
2003
Hemoglobin g/dl
16
14
12
10
8
6
4
2
0
Patient 1
2003 12.5 10.7 12 9.2
9.98
10.3
11.7
13.1 14.4
13.2 14.8
12.6
12.5
11.3
12.5
13.8 11.7 13
13 11.4
12.2
10.8
13.3
12.2
12.3 13.9
12.2
10.8
10.8
12.6
12
14.3 2004
2005
2006
2007
2008
2009
2010
Patient 2 Patient 3 Patient 4 Patient 5 2004 2005 2006 2007 2008 2009 2010
Figure3–DistributionofhemoglobinlevelsinpatientswithGaucherdiseasebetweentheyears2003and2010.
Discussion
Gaucherdiseaseispan-ethnic,butwithahighincidencein thepopulationofAshkenaziJews,involving1:450livebirths comparedto just 1:40.000inthe generalpopulation.7 Four alleles(N370S,L444P,84GG,IVS2+1)accountformostofthe mutationsthatcausethedisease,inparticularinAshkenazi Jewsduetothetraditionofmarryingwithinthesameethnic group.7
Thisstudyfocusesonararehealthproblemofonefamily inMaranhão.Accordingtoreports,thereisaverypertinent relationshiptotheoriginofthisfamilyconfirmedbythe mem-berswhoreportedJewishancestry,butdeniedanyrelationship betweentheparents.7
ThediagnosisofGDshouldbebasedonthemeasurement oftheglucocerebrosidaseenzymeinthepresenceof sugges-tiveclinical manifestationsofthedisease.When this does notconfirmdiagnosis,othermethodsareused,suchashigh levelsofchitotriosidaseenzymeandfamilymedicalhistory, bonemarrowbiopsy,molecularanalysis (genotyping), non-specificimagingresultsandlaboratoryexams.19Inthisfamily, fourmembers(36.3%)hadglucocerebrosidaseactivity sugges-tiveofGD,four(36.3%)levelssuggestingheterozygosityand two(18.1%)caseshadnormalenzymeactivity.Family mem-berII3, despite nothaving enzymatic values between0 to 1.9nmol/h/mgprotein,hadconfirmedGDdiagnosis.
Thereisastrongrelationshipbetweenthesemutationsand clinicalmanifestationsofthedisease.Thus,thegenotypecan helppredictseverityinpatients.Forinstance,homozygosity fortheN370Salleleisassociatedwithalesssevere pheno-type,althoughwithwideclinicalvariety.Heterozygosityfor N370SisprotectiveagainstimpairmentoftheSNC.19The fam-ilymembers withGDwere II1,II3,II4,II6,II8, allofwhom
hadtheN370S/?genotype(compoundheterozygote).Family member II2isfemale,heterozygouswiththeN/?genotype. The other individuals (II5,II7and II9) are normal, withno significantchangesintheirenzymelevels.
AnanalysisoftheprobabilityofindividualshavingGDin afamilywhoseparentsareheterozygousshowsthat25%of birthsarecompoundheterozygous(affectedbyGD),50% het-erozygous(oneallelewithamutationoftheGBAgene)and 25%arenormal.Inthiscase,ofthetotalofninechildren,five (55.5%)arecompoundheterozygotesforGD,thusshowingthe peculiarityofthiscase.
In a study conducted in Maranhão, 13 in a total of 14 patientswithGDhaddocumentedgenotyping.TheN370S/? genotypewasfoundin42.2%ofcases,whereasthepresence oftheN370Smutationwasidentifiedinmorethantwothirds ofthepatientsstudied.15InastudyfromSantaCatarinaState, theN370Sallelewasidentifiedin60%ofcases,with homozy-gosityinonlyonecase.TheL444Pmutantallelewaspresent inheterozygosiswiththeN370Smutationin20%ofpatients.7 GDisinheritedinanautosomalpatternandcanaffectboth genders.WithatotaloffivepatientsaffectedbyGDandall members of the same family,inthis study therewas pre-dominance offemalepatients (80%).In Maranhão,females are predominant in 64.3% of cases.15 In one of the most importantstudies onGD, therewas aslightpredominance offemalesat54%.20InBrazil,outofthetotalof551patients registeredandaccompaniedbytheGaucherRegistry,58%of patientsarefemale.Thesituationissimilarworldwide,with 53%femalesand47%malesofatotalof4764patientsenrolled inregistries.21
ERT was introduced in 1991 and has been shown to reverse or ameliorate many of the visceral and hema-tological manifestations of GD. ERT also improves bone manifestations,includingbonecrises,bonepain,and appear-ance of new osteonecrosis in the joints and pathological fractures, and improves bone mineral density to varying degrees.23
Aftermeasuring enzymelevels and genotyping, all five patientsaffected bythe diseaseinthis family fallinto the clinicalclassificationoftype1GD.Studiesreportthatmost patientsaretype1(94–96%type1),less than1%aretype2 andabout5%aretype3.8,24
Anemia has been reported in the literature in between 50%15and100%7ofcases.IntheInternationalCollaborative GaucherGroup(ICGG)report,anemiawaspresentin52%of casesbeforetreatmentwithimiglucerase.21 Inthestudy in SantaCatarinaState,leukopeniawasalsopresentin50%of cases.7 Inan Italianstudy,hemoglobin, and leukocyte and plateletcountsremainedstableafterERT.25Inseveral stud-ies thrombocytopenia was present: therewas a significant percentage(50%ofcases)inMaranhão.15Inonestudy, throm-bocytopeniawasfoundin56.25%of128patients,26partially disagreeingtodatafoundinthisstudy.
Hepatosplenomegalywasthemostcommonclinical fea-tureobservedinpatientswithGDinthecurrentstudy.Ina studyinMaranhão,hepatomegalyandsplenomegalyaffected 66.6%and83.3%ofpatients,respectively.15Inanotherstudy, hepatosplenomegalywasthemostcommonpathological find-ingatphysicalexamination(80%ofcases).7
There is a wide spectrum of skeletal complications related to GD, ranging from asymptomatic osteopenia to osteonecrosis, with secondary articular degeneration.27 A studyconductedinthestateofSantaCatarina,detectedbone abnormalitiesin75%ofpatientswiththemostcommonbeing osteopenia in40% ofcases,followed byavascular necrosis in30%and Erlenmeyerflaskdeformity in20%.7 In astudy conductedinSpain,40% ofpatients complained ofdiffuse bonepain.28InastudycarriedoutinIsrael,afterfouryears ofERT,therewas asignificant improvementinspleenand livervolumes,hemoglobinandplateletcountsandZ-scores forlumbarspineandfemur.23
BeforestartingtheERT,80%ofpatientshadabodymass index (BMI) between 18.5 and 24.9kg/m2. Onlyone of the
patients had a BMI between 25 and 29.9kg/m2. However,
in2010,sevenyearsafterstartingthe ERT,60% ofpatients enteredtheoverweightthresholdand40%hadnormalBMIs. Amongthose considered normal, onlyonelost body mass (Patient#5–from27.6kg/m2to23.8kg/m2).
Inconclusion,fivemembersofthisfamilyhadGDandwere followedupatHEMOMAR.Theageatdiagnosisand classifica-tionoftheGDtypecorrelatedtocasesregisteredintheState ofMaranhãowiththeprevalenceoftheN370Smutation.ERT improved hematological indexes, hepatosplenomegaly and thequalityoflifeofthesepatients.
Conflicts
of
interest
Theauthorsdeclarenoconflictsofinterest.
r
e
f
e
r
e
n
c
e
s
1.NathanDG,OskiFA.Storagediseasesofthereticuloendotelial system:Gaucher’sdisease.In:NathanDG,OskiFA,editors. NathanandOski’shematologyofinfancyandchildhood.5th ed.Philadelphia:WBSaunders;1998.p.1474–8.
2.BeautlerE,GrabowskiGA.Gaucherdisease.In:ScriverCR, BeaudetAL,SlyWS,ValleD,editors.Themetabolicand molecularbasesofinheriteddisease.8thed.NewYork: McGraw-Hill;2001.p.3635–68.
3.PetroianuA.Cirurgiasconservadorasdobac¸opara tratamentodaDoenc¸adeGaucher.RevBrasHematol Hemoter.2004;26(1):13–8.
4.KrugBC.Avaliac¸ãodaimplementac¸ãodoprotocoloclínicoe diretrizesterapêuticasdoMinistériodaSaúdeparaDoenc¸a deGauchernocentrodereferênciaestadual:impactosobre ospacientesesobreoSistemaÚnicodeSaúde.Dissertac¸ão (MestradoemCiênciasMédicas).PortoAlegre:Universidade FederaldoRioGrandedoSul;2007.
5.MaasM,HangartnerT,MarianiG,McHughK,MooreS, GrabowskiGA,etal.Recommendationsfortheassessment andmonitoringofskeletalmanifestationsinchildrenwith Gaucherdisease.SkeletalRadiol.2008;37(3):185–8.
6.LopesAraújoOliveiraMC,OliveiraBM,QueirósE,VianaMB. AspectosclínicosenutricionaisdaDoenc¸adeGaucher: estudoprospectivode13crianc¸asemumúnicocentro.J Pediatr.2002;78(6):517–22.
7.PaesCavalcantiFerreiraVL.EstudodaDoenc¸adeGaucherem SantaCatarina.Dissertac¸ão(MestradoemCiênciasMédicas). Florianópolis:UniversidadeFederaldeSantaCatarina;2003.
8.TrindadeeSilvaLP,SilvaH,CabreraH.Doenc¸adeGaucher. ActaMedPort.2007;20:175–8.
9.MartinsAM,LoboCL,SobreiraEA,ValadaresER,PortaG, SemionatoFilhoJ,etal.TratamentodaDoenc¸adeGaucher: umconsensobrasileiro.RevBrasHematolHemoter. 2003;25(2):89–95.
10.Michellin-TirelliK.Estudodascaracterísticasbioquímicasda
-glicosidasehumanaemindivíduoshomozigotose heterozigotosparaaDoenc¸adeGauchercommutac¸ões pré-determinadas:comparac¸ãocomindivíduosnormais. Tese(DoutoradoemCiênciasBiológicas).PortoAlegre: UniversidadeFederaldoRioGrandedoSul;2005.
11.Brasil.MinistériodaSaúde.PortariaSAS/MSn◦449,de08de
julhode2002.Doenc¸adeGaucher:protocoloclínicoe diretrizesterapêuticas.Brasília,DF;2002.
12.BrautbarA,ElsteinD,PinesB,KrienenN,HemmerJ,Buskila D,etal.FibramyalgiaandGaucher’sdisease.QJM.
2006;99(2):103–7.
13.QueirozCamposAraújoAP.Doenc¸asmetabólicascom manifestac¸õespsiquiátricas.RevPsiqClin.2004;31(6): 285–9.
14.ElsteinD,ZimranA.Reviewofthesafetyandefficacyof imiglucerasetreatmentofGaucherdisease.Biologics. 2009;3(1):407–17.
15.OliveiraGS.PerfilclínicodospacientescomDoenc¸ade GauchernoEstadodoMaranhão.Monografia(Ciências Médicas).SãoLuís:UniversidadeFederaldoMaranhão;2008.
16.BarrosLD,XimenesVS,RibeiroEM,SilvaVC.Estudode pacientescomDoenc¸adeGaucheremumhospitalterciário infantildeFortaleza.CearáRevPediatr.2008;9(1):30–7.
17.GEPLAN.GerênciadePlanejamentoeDesenvolvimento Econômico.SãoLuís:AtlasdoMaranhão;2002.
18.IBGE.AnuárioEstatísticodoBrasil.RiodeJaneiro:Instituto BrasileirodeGeografiaeEstatística;2009.
20.CharrowJ,AnderssonHC,KaplanP,KolodnyEH,MistryP, PastoresG,etal.TheGaucherRegistry:demographicsand diseasecharacteristicsof1698patientswithGaucherdisease. ArchInternMed.2000;160(18):2835–43.
21.ICGG.RelatórioAnualde2010.SãoPaulo:Gaucher Registry-Brasil;2010.
22.SobreiraEA,BrunieraP.Avaliac¸ãodedoisanosdetratamento daDoenc¸adeGauchertipo1comterapiadereposic¸ão enzimáticaempacientesdoestadodeSãoPaulo,Brasil.Rev BrasHematolHemoter.2008;30(3):193–201.
23.TukanI,Hadas-HapernI,AltarescuG,AbrahamovA,Einstein D,ZimranA.Achievementoftherapeuticgoalswithlow-dose imigluceraseinGaucherdisease:asingle-centerexperience. AdvHematol.2013:151506.
24.KaplanP,AnderssonHC,KacenaKA,YeeJD.Theclinicaland demographiccharacteristicsofnonneuronopathicGaucher
diseasein887childrenatdiagnosis.ArchPediatrAdolesc Med.2006;160(6):603–8.
25.DeromaL,SechiA,DardisA,MacorD,LivaG,CianaG,etal. Didthetemporaryshortageinsupplyofimiglucerasehave clinicalconsequences?RetrospectiveObservationalStudyon 34ItalianGauchertypeIpatients.JIMDRep.2013;7(1):117–22.
26.MankinHJ,TrahanCA,BarnettNA,LaugheadJ,BoveCM, PastoresGM.Aquestionnairestudyfor128patientswith Gaucherdisease.ClinGenet.2006;69(3):209–17.
27.PastoresGM,PatelMJ,FiroozniaH.Boneandjoint complicationsrelatedtoGaucherdisease.CurrRheumatol Rep.2000;2(2):175–80.