www.jped.com.br
ORIGINAL
ARTICLE
Whole-exome
sequencing
as
a
diagnostic
tool
for
distal
renal
tubular
acidosis
夽
Paula
Cristina
Barros
Pereira
a,
Flávia
Medeiros
Melo
a,
Luiz
Armando
Cunha
De
Marco
a,b,
Eduardo
Araújo
Oliveira
a,c,
Débora
Marques
Miranda
a,c,
Ana
Cristina
Simões
e
Silva
a,c,∗aInstitutoNacionaldeCiênciaeTecnologia---MedicinaMolecular(INCT-MM),UniversidadeFederaldeMinasGerais(UFMG),Belo
Horizonte,MG,Brazil
bDepartmentofSurgery,FacultyofMedicine,UniversidadeFederaldeMinasGerais(UFMG),BeloHorizonte,MG,Brazil cDepartmentofPediatrics,UnitofPediatricNephrology,InterdisciplinaryLaboratoryofMedicalInvestigation,Facultyof
Medicine,UniversidadeFederaldeMinasGerais(UFMG),BeloHorizonte,MG,Brazil
Received11November2014;accepted25February2015 Availableonline22July2015
KEYWORDS ATP6V0A4; ATP6V1B1; Children;
Distalrenaltubular acidosis;
Genetics; Whole-exome sequencing
Abstract
Objective: Distalrenaltubularacidosis (dRTA)ischaracterized bymetabolicacidosis dueto impairedrenalacidexcretion.Theaimofthisstudywastodemonstratethegeneticdiagnosis offourchildrenwithdRTAthroughuseofwhole-exomesequencing.
Methods: Twounrelatedfamilieswereselected;atotaloffourchildrenwithdRTAandtheir parents,inordertoperformwhole-exomesequencing.Hearingwaspreservedinbothchildren fromthefirstfamily,butnotinthesecond,whereinatwinpairhadseveredeafness. Whole-exomesequencing was performedintwo pooled samples andfindings were confirmedwith Sangersequencingmethod.
Results: TwomutationswereidentifiedintheATP6V0A4andATP6V1B1genes.Inthefirst fam-ily,anovelmutationintheexon13 oftheATP6V0A4genewith asinglenucleotidechange GAC→TAC (c.1232G>T)was found,which causedasubstitutionofaspartic acidtotyrosine inposition 411.In thesecond family,ahomozygousrecurrent mutationwithone base-pair insertion(c.11491155insC)inexon12oftheATP6V1B1genewasdetected.
Conclusion: Theseresultsconfirmthevalueofwhole-exomesequencingforthestudyofrare andcomplexgeneticnephropathies,allowingtheidentificationofnovelandrecurrent muta-tions.Furthermore,forthefirsttimetheapplicationofthismolecularmethodinrenaltubular diseaseshasbeenclearlydemonstrated.
©2015SociedadeBrasileiradePediatria.PublishedbyElsevierEditoraLtda.Allrightsreserved.
夽
Pleasecitethisarticleas:PereiraPC,MeloFM,DeMarcoLA,OliveiraEA,MirandaDM,SimõeseSilvaAC.Whole-exomesequencingasa diagnostictoolfordistalrenaltubularacidosis.JPediatr(RioJ).2015;91:583---9.
∗Correspondingauthor.
E-mail:[email protected](A.C.SimõeseSilva). http://dx.doi.org/10.1016/j.jped.2015.02.002
PALAVRAS-CHAVE ATP6V0A4;
ATP6V1B1; Crianc¸as;
Acidosetubularrenal distal;
Genética;
Sequenciamentototal doexoma
Sequenciamentototaldoexomacomoferramentadediagnósticodeacidosetubular renaldistal
Resumo
Objetivo: Aacidosetubularrenaldistal(ATRd)écaracterizadaporacidosemetabólicadevido aexcrec¸ãorenal de ácido prejudicada.O objetivo desteartigo é apresentar o diagnóstico genéticodequatrocrianc¸ascomATRdutilizandoosequenciamentototaldoexoma.
Métodos: Selecionamosduasfamíliasnãorelacionadas,totalizandoquatrocrianc¸ascomATRd eseuspais,pararealizarosequenciamentototaldoexoma.Aaudic¸ãofoipreservadaemambas ascrianc¸asdafamíliaum,porémemnenhumacrianc¸adafamíliadois,naqualumpardegêmeas teveperdaauditivasevera.Realizamososequenciamentototaldoexomaemdoisconjuntosde amostraseconfirmamososachadoscomométododeSequenciamentodeSanger.
Resultados: Duasmutac¸õesforamidentificadasnosgenes ATP6V0A4eATP6V1B1.Nafamília um,detectamosumanovamutac¸ãonoéxon13dogeneATP6V0A4comumaalterac¸ãoemum nucleotídeo únicoGAC→ TAC (c.1232G>T)que causousubstituic¸ãode ácido aspártico por tirosinanaposic¸ão411.Nafamíliadois,detectamosumamutac¸ãorecorrentedohomozigoto cominserc¸ãodeumpardebases(c.11491155insC)noéxon12dogeneATP6V1B1.
Conclusão: Nossosresultados confirmamo valor dosequenciamento total doexoma para o estudodenefropatiasgenéticascomplexas,permitindoaidentificac¸ãodemutac¸õesnovase recorrentes.Adicionalmente,demonstramosclaramentepela primeiravezaaplicac¸ãodesse métodomolecularemdoenc¸astubularesrenais.
©2015SociedadeBrasileiradePediatria.PublicadoporElsevierEditoraLtda.Todososdireitos reservados.
Introduction
Distal renal tubular acidosis (dRTA) is a rare and com-plex renal disease due to a defect in the excretion of acid load (H+ and ammonium ions) in alpha-intercalated cells of the collecting duct. The acid load accumulation in the distal nephron results in consumption and reduc-tion of the bicarbonate/CO2 buffer in blood.1 The main
clinical features of dRTA are vomiting, diarrhea, and/or
constipation, loss of appetite, polydipsia, and polyuria.
Chronicacidosisandsecondaryalterationssuchasvomiting,
polyuria, and dehydration affect growth, leading to
fail-uretothrive.Ultrasoundstudiescanshownephrocalcinosis
and/ornephrolithiasis.2Ingeneral,dRTAhasgoodprognosis
ifitisdiagnosedatan earlyageandalkaline treatmentis
continued.Untreated,dRTAcausesgrowthretardationand
ricketsinchildrenandosteomalaciainadults.Deterioration
ofrenalfunctioncanoccurovertheyears.3
Distal RTA can be transmitted as either an autosomal
dominantoran autosomalrecessivetrait.4 Theautosomal
dominantphenotypetypicallycoursesmildlyinadolescence
oradulthood;4oneparentsuffersfromandisthecarrierof
thedisease,oritisduetodenovomutation.Mutationsinthe
SLC4A1geneinfamilieswithautosomaldominantdRTAhave
beenidentified.2,5,6 Thesymptomsintheautosomal
reces-sive phenotype predominantly appear at infancy or early
childhood,inwhichgrowthretardationisverycommon.This
variantcanoccurwithorwithoutdeafness,andparentsare
notaffected.2AutosomalrecessivedRTAisassociatedwith
mutationsinanyofthefollowinggenes:SLC4A1,7ATP6V0A4,
andATP6V1B1.2,8 Individuals without hearing defects
usu-allycarrymutationsintheATP6V0A4gene,whilethosewith
deafnesshaveATP6V1B1genemutations.Inapproximately
20%ofthepatientswithdRTA,nomutationswerefoundin
anyoftheserelatedgenes.3Indeed,therearedRTApatients
withdeafnesswithoutATP6V1B1 genemutations,and
oth-ers withnormal hearing whodonot have ATP6V0A4 gene
mutations.3 Thesefindings suggestthatother transporters
orchannelsmightcausedRTA.Intermsofcomplexity,itis
knownthatsomepatientswithmutationsintheATP6V0A4
gene develop deafness only in the second decade of life.
Thus, there remains much to be elucidated in terms of
phenotype---genotypecorrelations.8---10 Sofar,morethan20
mutationsinATP6V0A4arealreadyknown.
Whole-exome sequencing provides coverage of more
than95%oftheexons,whichcontain85%ofdisease-causing
mutations in Mendelian disorders and many
disease-predisposing single nucleotide polymorphisms (SNPs)
throughout the genome.11,12 Whole-exome sequencing is
worthwhile to evaluate the disease pathogenesis and to
recognize new pathogenic genes or mutations associated
to disorder, especially in Mendelian disorders.11,12 In this
regard,thepresentstudyaimedtoevaluatetheusefulness
ofwhole-exomesequencingforgeneticdiagnosisofdRTA.
Patients
and
methods
Subjectsandclinicalassessment
(two girls), diagnosed withdRTA andnerve deafness, and their healthy mother;the father is unknown. Allpatients weresubmittedtoasystematicprotocol includingclinical andnutritionalevaluation,laboratorymeasurements,renal ultrasonography, and genetic analysis. Informed consent, approved by the institutional Ethics Board of UFMG, was obtained fromall participants; in thecase of children,it wasalsoobtainedfromtheirparent/guardian.
DNAextraction
GenomicDNAwasextractedfrom5mLofdRTApatientsand theirparent’speripheralblood,usingaQIAampBloodDNA mini Kit (Qiagen®, Milano, Italy) according to the manu-facturer’sinstructions.Allsampleswerequalitycontrolled for purity using a NanoDrop spectrophotometer (Thermo Scientific®, Waltham, USA). DNA samples were stored at −20◦Cuntilusage.
Whole-exomesequencing
Exomesequencingwasperformedontwopoolsofsamplesto optimizetheresults.Sampleswerepooledconsidering the clinicalfeaturesofthepatients.ThefirstpoolhadDNAfrom thetwosiblingswithdRTAwithoutdeafness,andthesecond, fromthetwinsisters withdRTAassociatedwithdeafness. Arraycapturewasusedtoisolatetherelevanthumangenes (SeqCap EZ Human Exome Library v2.0, Roche®, Basel, Switzerland) andthesegenesweresequenced onthe Illu-mina HiSeq 2000plataform (Sigma---Aldrich Corporation®, Missouri,USA).
Filteringdata
The following principle stepswere takentoprioritize the high-qualityvariants:(i)variantswithinintergenic,intronic, and untranslated region (UTR) regions and synonymous mutations were excluded from downstream analysis; (ii) variants with quality score less than 20 were excluded; (iii) onlytheconservationscore(phyloP)fromcomparison of human and 43 vertebrates higher than 3 were consid-ered; (iv) after this prior selection, the remaining genes were filtered by the function. The software PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/)predicted
possi-bleimpactofvariants.Thefinalsetofselectedvariantswas
visuallyinspected usinganIntegrative GenomicsViewer.13
Previously described polymorphic variants in public data
wereinvestigatedandcomparedwiththevariationsfound
inthecurrentexome.Theselectedmutationstobe
investi-gatedineachgroupofthisstudywerenotfoundinprevious
exomesequences(http://evs.gs.washington.edu/EVS/).
Validationofdata
Polymerase chain reaction (PCR) Sanger sequencing was used in the analysis to confirm the data. All patients and their parents were submitted to the PCR. Amplifi-cation products of appropriate size were identified using polyacrylamide gel electrophoresis. Products were puri-fied using the QIAquick PCR purification kit (Qiagen®,
Milano, Italy) and then submitted to sequencing reac-tionusingboth forward andreverse primers withthe ABI BigDye Terminator Cycle Sequencing Kit v. 3.1 on an ABI PRISM3730XL GeneticAnalyzer(Applied Biosystems®, Fos-ter City, USA). Each read was aligned to the reference sequence, and mutations were identified with Sequencer software (http://www.genecodes.com). All primers were
designedusingtheonlinetoolPrimer3.Theprimersforexon
12of ATP6V1B1gene were:5′TTGACCCCTCGGAATGTAGG3′
and 5′CCGGACCCTCTTCTCCTTAC3′ (product size of 238
base pairs). The primers for exon 13 of ATP6V1B1 were:
5′ATGCAAATCGTGGAGCTGTG3′ and 5′
ATGAATCAGGGCAA-GACGGT3′(productsizeof264basepairs).
Structuralstudiesofmutations
ProteinandDNAsequencealignmentswereperformedusing the ClustalW and the MultAlin (http://multalin.toulouse. inra.fr/multalin/), respectively. The prediction of amino
acid substitution on the biological function of the
pro-tein was evaluated using both PolyPhen-2 and Provean
software (http://provean.jcvi.org and http://genetics.
bwh.harvard.edu/pph2/,respectively).
Results
dRTAwithoutdeafness
Thisfamilyconsistedoftwosiblings,a13-year-oldgirland her7-year-oldbrother,withwell-defineddRTA.Thegirlwas theproband,diagnosedwithdRTAattheageof4months. The initialfindings werefailure tothrive,hyperchloremic metabolicacidosiswithabnormallyhighurinepH(7.0), nor-mal venous blood pH (7.36), normal glomerular filtration rate,andnephrocalciosis.Theboywasdiagnosedinhis1st month of life, after a severe dehydration withmetabolic acidosis, hypokalemia, transient elevation of serum cre-atinine,andhypocalcemia. His firstrenalultrasonography showedbilateralnephrocalcinosis.Table1summarizes
clin-icalandbiochemicalmanifestationsat baselineleadingto
the diagnosis of dRTA in each patient. The parents were
unaffected,andhadanolderchildthatdiedbytheageof4
monthswithsimilarsymptoms.
Whole-exomesequencingconductedinthisgroup
gener-ated3577singlenucleotidevariations(SNVs)and416small
insertionsanddeletions(INDELs).Filteringforvariantswas
appliedtoselectthecandidategene(Table2).
Afterfilteringtheexomedata,theATP6V0A4genewas
selectedfor study.A singlenucleotidechange GAC→TAC
(c.1232G>T) in exon 13 was observed, which caused an
aminoacid substitution: aspartic acid totyrosine in
posi-tion411(p.D411Y). Thisaminoacidchangewaspredicted
tobedamaging byProveanandPolyPhen-2.This mutation
occursatanevolutionarilyconservedaminoacidandaffects
highlypreservedresidues(datanotshown).
ThepatientsandtheirparentsweresubmittedtoSanger
sequencingbyusingthedesignedprimerforexon13ofthe
ATP6V0A4gene.Thetwosiblingspresentedthesame
muta-tioninhomozygosis(c.1232G>T),whilebothparentshada
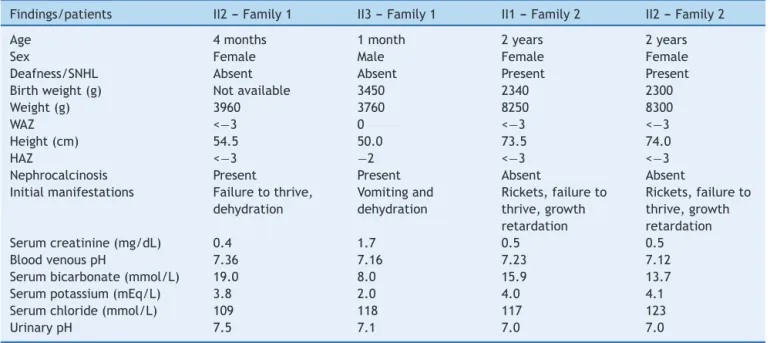
heterozygoustrace(Fig.1AandB).Thisisanovelautosomal
Table1 ClinicalandbiochemicalfindingsatbaselineofpatientswithdRTA.
Findings/patients II2---Family1 II3---Family1 II1---Family2 II2---Family2
Age 4months 1month 2years 2years
Sex Female Male Female Female
Deafness/SNHL Absent Absent Present Present
Birthweight(g) Notavailable 3450 2340 2300
Weight(g) 3960 3760 8250 8300
WAZ <−3 0 <−3 <−3
Height(cm) 54.5 50.0 73.5 74.0
HAZ <−3 −2 <−3 <−3
Nephrocalcinosis Present Present Absent Absent
Initialmanifestations Failuretothrive, dehydration
Vomitingand dehydration
Rickets,failureto thrive,growth retardation
Rickets,failureto thrive,growth retardation
Serumcreatinine(mg/dL) 0.4 1.7 0.5 0.5
BloodvenouspH 7.36 7.16 7.23 7.12
Serumbicarbonate(mmol/L) 19.0 8.0 15.9 13.7
Serumpotassium(mEq/L) 3.8 2.0 4.0 4.1
Serumchloride(mmol/L) 109 118 117 123
UrinarypH 7.5 7.1 7.0 7.0
Romannumeralsindicatethefamilypositiononthepedigree:II2,Family1referstothesecondproband(daughter)offamilyone;II3, Family1referstothethirdproband(son)offamilyone;II1,Family2---thefirsttwindaughteroffamilytwo;II2,Family2---thesecond twindaughter;SNHL,sensorineuralhearingloss;WAZ,weight-for-agez-score;HAZ,height-for-agez-score.
dRTAwithdeafness
This family consisted of a twin pair of girls with dRTA in associationwithnervedeafness. The girlswerediagnosed attheageof2afteralongperiodoftreatingforricketsand growth retardation with only nutritional support. Clinical andbiochemicalfeaturesatbaselineareshowninTable1.
Whole-exome sequencing conducted in the family two
generated 4375 SNVs and 2416 INDELs. After filteringthe
variants,onlyonecandidategeneremained(Table2).
Based on the exome data, the ATP6V1B1 gene was
selected as the candidate in this group. A homozygous
one base-pair insertion (c.11491155insC) in exon 12 was
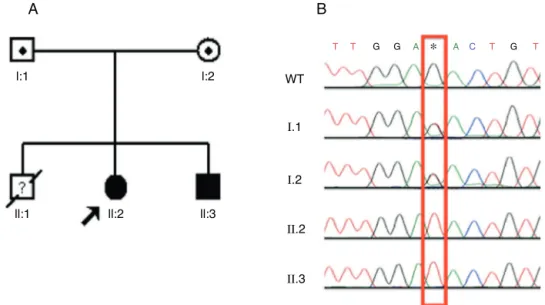
detected (Fig. 2A). The two dRTA twin sisters presented
the insertion described. PCR of their unaffected mother
wasperformed(fatherunknown).Thetwosiblingshadthe
same homozygous mutation, while the mother presented
thisinsertioninheterozygosis(Fig.2BandC).
Discussion
Several DNA errorsare located in exons leadingto struc-tural alterations in proteins and functional changes.3 In
thismanner, whole-exomeanalyzes theseexonsin arapid
and cost-effectivemanner, allowing geneticevaluation of
complexandmonogenicdiseases.14,15Inraredisorders,the
useofwhole-exomemayminimizethefailureindetecting
mutationsathot-spotsregions.Nonetheless,directSanger
sequencingisstillconsideredthemostaccuratemethodto
findmutations,sinceothergenetestingtechniques,suchas
wholeexome,maynotdetectallsequencevariations.The
search ina whole-genome basisand validationof findings
withSangersequencingmethodappears tobeanefficient
waytodeterminegeneticcausalityofadisorder.However,
the incorporation of these next-generation technologies
intoclinical practiceisstillchallenging,sincethese
tech-niquesareremainveryexpensive,anddatainterpretation
Table2 VariantprioritizingforFamily1andforFamily2.
Parameters Family1 Family2
Numberofvariants Numberofvariants
Totalvariants 3993 6791
Intergenic,intronic,andUTRregionsand synonymousmutationswereexcluded
1445 1912
Qualityscore<20wereexcluded 879 1012
PhyloP<3wereexcluded 131 215
Selectionbyfunction 15 14
PolyPhen<0.7wereexcluded 10 6
Selectionusingcross-referencegenedatabaseand IGV
1 1
Candidategene 1 1
A
l:1
ll:1 ll:2 ll:3
l:2 WT
T T G G T T
∗
GA A C
I.1
I.2
II.2
II.3
B
Figure1 Identification,pedigreeofFamily1,andresultsofsequencingfor c.1232G>Tmutation.(A)Thepedigreeshowsthe affectedstatuses,individualidentifiers,andgenotypesatcodon411.Thearrowindicatestheproband.(B)DNAsequence chro-matogramsinwhichthetwoaffectedsiblingshavehomozygousGtoTsubstitutionatc.1232.Thissubstitutionoccursinheterozygosis inbothparents.WT,wildtype.*Mutatednucleotide.
is laborious and difficult.11,12 Recent studies suggest that
whole-exomesequencingwould beuseful toevaluate
dis-easepathogenesisandtorecognizenewpathogenicgenes
or mutations associated to disorder, special in Mendelian
disorders.16 Thus, the present study used whole exome
followed by Sanger sequencing as a strategy for genetic
diagnosisofdRTAinfourchildren.
These results showed, for the first time, the utility
of whole-exome sequencing in renal tubular disorders,
by allowing the identification of a recurrent and a new
pathogenicmutationindRTA.InheritedformsofdRTAhave
threevariants:autosomal dominantand autosomal
reces-sive,withorwithoutdeafness.4Dominantdiseasetypically
presents more mildly in adolescence or adulthood, and
it has been only associated to mutations in the
bicar-bonate/chloride exchanger (AE1). However, the recessive
variantoccursininfancyorearlychildhood,inwhichgrowth
retardation is very common,4 asobserved in the present
cases.Autosomalrecessive dRTAhasbeen associatedwith
mutations in the genes ATP6V1B1 and ATP6V0A4, which
A
l:1
Allele1wt Allele2Ins Consensus
1145 1155 1164 1174
lI:1 lI:2
l:2
WT
T A AT A
C C C C C C C C C
I.2
II.1
II.2
B
C
encode the subunits a4 and B1 of the vacuolar-type pro-tonATPase(V-orH+-ATPase),respectively.17---22Inaddition,
mutationsintheSLC4A1gene,responsiblefor the
expres-sionof AE1proteins, have been alsodetected in casesof
autosomalrecessivedRTAwithoutdeafness.23---28
Indeed, mutations in different subunits of the proton
pumpthatareexpressedinkidneyandeartissuescancause
tubulardefects associatedwith deafness.17 The
vacuolar-type proton ATPase (V- or H+-ATPase) is a pump with
multiplesubunitsthatisessentialfor normalacidification.
Twostructuraldomainsformthis pump:membrane-bound
V0 and cytoplasmic or peripheral V1. Each domain
com-prisesmultiplesubunits(a---eandA---H,respectively),which
are responsible for ATP hydrolysis and proton transport,
respectively.4 The ATP6V1B1 gene encodes the B1
sub-unit, while the ATP6V0A4 gene encodes the a4 subunit.
The vacuolar-type proton ATPase is expressed apically on
renal␣-intercalatedcells,andinthecochleaand
endolym-phaticsac. Basedonthe typeofhearingloss,thesetypes
of mutations can be suspected. Conductive deafness was
observedin mutations of the intracellular isoformof
car-bonic anidrase (CA), whereas sensorineural hearing loss
(SNHL)hasbeen associatedwithATP6V1B1 andATP6V0A4
mutations.18---20 Accordingly, a homozygous one base-pair
insertion (c.1149 1155insC) was found in exon 12 of the
ATP6V1B1 gene in twin sisters with SNHL. Mutations in theSLC4A1geneusuallydonothave anyassociation with
deafness.22---28Therefore,thepresenceandthekindof
deaf-nessarehelpfulindistinguishingdifferentformsofdRTA.
Inthetwofamiliesofthisstudy,parentswereunaffected
and dRTA had early onset, leading togrowth impairment
during infancy. Therefore, mutations in the SLC4A1 gene
(AE1proteins)werehighlyunlikelyinthepatients.InFamily
1,exomesequencingidentifiedanovel homozygous
muta-tion in the ATP6V0A4 gene. Based on previous reports10
andontheclinicalandbiochemicalfeatures,thisgenewas
selectedasapotentialcandidatetosearchformutations,
sincenohearinglosswasdetectedintheaffectedpatients.
A singlenucleotide change in exon13 wasobserved that
caused an amino acid substitution: aspartic acid to
tyro-sineinposition411.Thisaminoacidchange waspredicted
tobedamagingbyProveanandPolyPhen-2.Unfortunately,
nofunctionalstudieswereperformedtodecipherthe
pre-ciseroleofthismutation.However,itshouldbementioned
that this mutation occurs at an evolutionarily conserved
aminoacidand affects highly preserved residues. In
addi-tion,thesubstitutionofasparticacidbytyrosinemaychange
chemicalproperties ofthe proteinat criticalregions. For
instance,thischangemayaltertheisoelectricpointofthe
protein,considering that tyrosine is a neutral aminoacid,
whileasparticacidisanacid.
The twingirls of family twohadSNHL andearly-onset
symptoms of dRTA. The phenotypes, together with the
whole-exome data, led the authors to investigate the
ATP6V1B1gene.Accordingly,a previouslydescribed
muta-tion was found,9 which was also confirmed by Sanger
sequencing.Itshouldbepointed,however,thatATP6V1B1or
ATP6V0A4genemutationshavenotbeennotfoundinsome
familieswith primaryrecessive forms of dRTA. There are
numerousothercandidategenesforrecessivedRTA,29
espe-ciallythosegenesrelatedtotheprotontransporters.Inthis
regard,theuseof whole-exomesequencingtogetherwith
thephenotypecharacteristicsmayresultinthediscoveryof
newmutationsandgeneticalterationsinthiscomplexand
raredisease.
Insummary,whole-exomesequencingfollowedbySanger
sequencingwasasuccessfulstrategyinidentifyingnoveland
recurrentmutationsinthesecasesofdRTA.However,which
geneticvariantsarepotentially causativeof renaltubular
transportalterationsremainstobeelucidated,especiallyin
recessiveformsdRTA.
Conflicts
of
interest
Theauthorsdeclarenoconflictsofinterest.
Acknowledgements
This study was partially supported by CNPq (Conselho Nacional de Desenvolvimento Científico e Tecnológico, Brazil) and FAPEMIG (Fundac¸ão de Amparo à Pesquisa do EstadodeMinasGerais,Brazil),bytheGrantINCT-MM (Insti-tutoNacionaldeCiênciaeTecnologia---MedicinaMolecular: FAPEMIG:CBB-APQ-00075-09/CNPq573646/2008-2). Dr.LA DeMarco,Dr.EAOliveira,Dr.DMMiranda,andDr.ACSimões eSilvareceivedaresearchgrantfromCNPq.
References
1.RodríguezSorianoJ.Renaltubularacidosis:theclinicalentity. JAmSocNephrol.2002;13:2160---70.
2.Fry AC, Karet FE. Inherited renal acidoses. Physiology (Bethesda).2007;22:202---11.
3.EscobarL,MejíaN,GilH,SantosF.Distalrenaltubularacidosis: ahereditarydiseasewithaninadequateurinaryH+excretion.
Nefrologia.2013;33:289---96.
4.Pereira PC, Miranda DM, Oliveira EA, Silva AC. Molecular pathophysiology of renal tubular acidosis. Curr Genomics. 2009;10:51---9.
5.BruceLJ,CopeDL,JonesGK,SchofieldAE,BurleyM,PoveyS, etal.Familialdistal renaltubularacidosisisassociatedwith mutationsintheredcellanionexchanger(Band3,AE1)gene. JClinInvest.1997;100:1693---707.
6.Karet FE,Gainza FJ, GyöryAZ, Unwin RJ, Wrong O, Tanner MJ, et al. Mutations in the chloride-bicarbonate exchanger geneAE1causeautosomaldominantbutnotautosomal reces-sivedistal renal tubularacidosis. Proc Natl Acad SciU SA. 1998;95:6337---42.
7.TanphaichitrVS,SumboonnanondaA,IdeguchiH,ShayakulC, BrugnaraC,TakaoM,etal.NovelAE1mutationsinrecessive distalrenaltubularacidosis.Loss-of-functionisrescuedby gly-cophorinA.JClinInvest.1998;102:2173---9.
8.Vargas-PoussouR,HouillierP,LePottierN,StrompfL,LoiratC, BaudouinV,etal.Geneticinvestigationofautosomalrecessive distalrenaltubularacidosis:evidenceforearlysensorineural hearinglossassociatedwithmutationsintheATP6V0A4gene.J AmSocNephrol.2006;17:1437---43.
9.KaretFE,FinbergKE,NelsonRD,NayirA,MocanH,SanjadSA, etal.MutationsinthegeneencodingB1subunitofH+-ATPase
causerenaltubularacidosiswithsensorineuraldeafness.Nat Genet.1999;21:84---90.
11.SmithA,BoycottKM,JarinovaO.LakeLouisemutation detec-tion meeting 2013: clinical translation of next-generation sequencingrequiresoptimizationofworkflowsand interpreta-tionofvariants.HumMutat.2014;35:265---9.
12.RabbaniB,TekinM,MahdiehN.Thepromiseofwhole-exome sequencinginmedicalgenetics.JHumGenet.2014;59:5---15. 13.RobinsonJT,ThorvaldsdóttirH,WincklerW,GuttmanM,Lander
ES,GetzG,etal.Integrativegenomicsviewer.NatBiotechnol. 2011;29:24---6.
14.Choi M, Scholl UI, Ji W, Liu T, Tikhonova IR, Zumbo P, et al. Genetic diagnosis bywhole exome capture and mas-sively parallel DNA sequencing. Proc Natl Acad Sci U S A. 2009;106:19096---101.
15.BonnefondA,DurandE,SandO,DeGraeveF,GallinaS,Busiah K,etal.Moleculardiagnosisofneonataldiabetesmellitususing next-generationsequencing of thewhole exome.PLoS ONE. 2010;5:e13630.
16.YangY,MuznyDM,ReidJG,BainbridgeMN,WillisA,WardPA, et al. Clinical whole-exomesequencing for thediagnosis of Mendeliandisorders.NEnglJMed.2013;369:1502---11. 17.LangF,VallonV,KnipperM,WangemannP.Functional
signifi-canceofchannelsandtransportersexpressedintheinnerear andkidney.AmJPhysiolCellPhysiol.2007;293:C1187---208. 18.StoverEH,BorthwickKJ,BavaliaC,EadyN,FritzDM,Rungroj
N,etal.NovelATP6V1B1andATP6V0A4mutationsinautosomal recessivedistalrenal tubularacidosiswithnewevidencefor hearingloss.JMedGenet.2002;39:796---803.
19.BatlleD,HaqueSK.Geneticcausesandmechanismsofdistal renaltubularacidosis.NephrolDialTranspl.2012;27:3691---704. 20.GilH,SantosF,GarcíaE,AlvarezMV,Ordó˜nezFA,MálagaS,etal. DistalRTA withnervedeafness: clinicalspectrumand muta-tionalanalysisinfivechildren.PediatrNephrol.2007;22:825---8.
21.MiuraK,SekineT,TakahashiK,TakitaJ,HaritaY,OhkiK,etal. Mutationalanalysesofthe ATP6V1B1and ATP6V0A4genes in patientswithprimarydistalrenaltubularacidosis.NephrolDial Transpl.2013;28:2123---30.
22.ElhayekD,PerezdeNanclaresG,ChouchaneS,HamamiS,Mlika A,TroudiM,etal.Moleculardiagnosisofdistalrenaltubular acidosisinTunisianpatients:proposedalgorithmforNorthern Africa populations for the ATP6V1B1, ATP6V0A4 and SCL4A1 genes.BMCMedGenet.2013;14:119.
23.AlperSL,DarmanRB,ChernovaMN,DahlNK.TheAEgenefamily ofCl/HCO3-exchangers.JNephrol.2002;15:S41---53.
24.SchusterVL.Functionandregulationofcollectingduct interca-latedcells.AnnuRevPhysiol.1993;55:267---88.
25.Chang YH, Shaw CF, Jian SH, Hsieh KH, Chiou YH, Lu PJ. Compoundmutationsinhumananionexchanger1are associ-atedwithcompletedistalrenaltubularacidosisandhereditary spherocytosis.KidneyInt.2009;76:774---83.
26.AlperSL.MolecularphysiologyandgeneticsofNa+-independent
SLC4anionexchangers.JExpBiol.2009;212:1672---83. 27.Cheidde L,Vieira TC,LimaPR,SaadST, HeilbergIP.Anovel
mutationintheanionexchanger1geneisassociatedwith famil-ialdistalrenaltubularacidosisandnephrocalcinosis.Pediatrics. 2003;112:1361---7.
28.Wrong O, Bruce LJ, Unwin RJ, Toye AM, Tanner MJ.Band 3 mutations,distal renal tubularacidosis, andSoutheast Asian ovalocytosis.KidneyInt.2002;62:10---9.