Serviço de Neurologia do Hospit al de Clínicas da Universidade Federal do Paraná, Curit iba, PR, Brazil (UFPR): 1Resident e em Neurologia;2M édico Neurologist a; 3Prof essor Assist ent e, 4Prof essor Adjunt o; 5Prof essor Tit ular; 6M édico Genet icist a, Laborat ório Genet ika, Curit iba - PR Brazil. 7Klinisch-Genet isch Cent rum Nijmegen, Nijmegen - Net herlands
Received 18 February 2004, received in f inal f orm 22 June 2004. Accept ed 3 August 2004.
Dra. Viviane F. Zét ola Serviço de Neurologia, Hospit al de Clínicas Rua General Carneiro 181 / sala 1236 80060900 Curit iba PR -Brasil. E-mail: viviane@f lumignano.com
Cerebrot endinous xant homat osis (CTX) is a rare aut osomal recessive disease charact erized by lipid accumulation in several tissues, mainly in the eye lses, cent ral nervous syst em (CNS) and muscle t en-dons. M ut at ions in t he gene t hat codes f or t he he-patic enzyme sterol 27-hydroxylase (CYP27A1) have been f ound in t hese pat ient s. We present t he f irst descript ion of a Brazilian f amily w it h suggest ive cli-nical presentation and diagnosis confirmation thro-ugh genet ic t est of CYP27A1 gene.
CASE
Case 1 – A 47 years old w hit e man w hich sympt oms
began at 20 years old (according to the mother’s informa-tion). He presented progressive walking difficulty,
becom-ing unable t o w alk f ast at 40 years old. Since t he begin-ning he presented cognitive difficulties and bilateral sw el-ling of Achilles t endons. He present ed delay of t he psy-chomot or development and st art ed w alking by one year and a half and he acquired t he capacit y t o run and t o go up st airw ays just at f ive years of age. The mot her ref er-red t hat he had a hist ory of chronic diarrhea during t he childhood. She denied ot her f amily members w it h simi-lar sympt oms, except f or t he pat ient ’s younger brot her.
Physical examination disclosed no abnormalities except f or bilat eral sw elling of Achilles t endons (Fig 1a). In t he neurological examination, he presented mild mental ret ar-dat ion, incomprehensive disart hria, mild bilat eral f acial palsy w it hout ot her f indings in cranial nerves. His mus-cular strength was IV - in superior limbs (SL) and IV- in infe-rior limbs (IL) according to Medical Research Council (M RC)
CEREBROTENDINOUS XANTHOM ATOSIS
Report of t w o Brazilian brot hers
M arcos Christ iano Lange
1, Viviane Flumignan Zét ola
2, Helio A.G. Teive
3,
Rosana H. Scola
4Ana Paula Trent in
2, Jorge A. Zavala
1, Eduardo R. Pereira
1,
Salmo Raskin
6, Lineu C. Werneck
5, Erik A. Sist ermans
7ABSTRACT - Cerebrot endinous xant homat osis is a t reat able rare aut ossomal recessive disease charact eri-zed by lipid st orage secondary t o a st erol 27-hydroxylase def iciency in t he f ormat ion of cholic and cheno-deoxycholic acids. We describe t w o Brazilian brot hers w it h cognit ive impairement and chronic diarrhea. One of t hem also present s bilat eral cat aract s. Neurological f indings w ere progressive w alking def icit , limb at axia and pyramidal signs. Bot h pat ient s had bilat eral Achilles t endon xant homat a. M agnet ic resonance i-mage showed signal alterations in cerebellar hemispheres. We describe these cases with molecular genetic analysis conf irming diagnosis and comparing w it h previous lit erat ure. The CYP27A1 gene st udy show ed a C1187T mut at ion on exon 6.
KEY WORDS: cerebrot endinous xant homat osis, st erol 27-hydroxylase, CYP27A1.
Xant om at ose cerebrot endínea: relat o de dois irm ãos brasileiros
RESUM O - Xant omat ose cerebrot endínea é doença aut ossômica recessiva t rat ável causada pelo acúmulo de lipídeos por deficiência da enzima 27-esterol hidroxilase na produção de ácido cólico e deoxicólico. Descreve-mos dois irmãos brasileiros com dif iculdade cognit iva e diarréia crônica. Um deles apresent ava cat arat a bi-lat eral. Os achados neurológicos f oram dif iculdade progressiva para deambular, at axia de membros e sinais piramidais. Ambos t inham xant omas de t endão aquileu bilat eralment e. O exame de ressonância magnét i-ca revelou áreas de sinal hiperintenso em ambos os hemisférios cerebelares. Descrevemos os i-casos com diagnós-tico genédiagnós-tico comparando-os com a literatura. O estudo do gene CYP27A1 demonstrou a mutação C1183T no exon 6.
Fig 1. Achilles t endon xant homas (arrow s): case 1 (A) and case 2 (B).
Fig 2. Brain M RI of t he case 1 (A) [coronal T2WI sequence] and case 2 (B) [axial ‘Flair’ sequence] show ed hiperint ense signal in bot h cerebellar hemispheres (arrow s).
rat ing. We not ed moderat e spast icit y in IL, preserved trophism and increased deep tendon reflexes with bilat e-ral Babinski sign. The sensibilit y w as preserved and t here w as ret ract ion in bilat eral Aquilles t endon. The pat ient present ed symmet rical dismet ry and disdiadococinesy in SL. The cerebellar clinical evaluat ion of IL w as disabled by the limitation of the movements. He had also gait atax-ia and w alked w it h support .
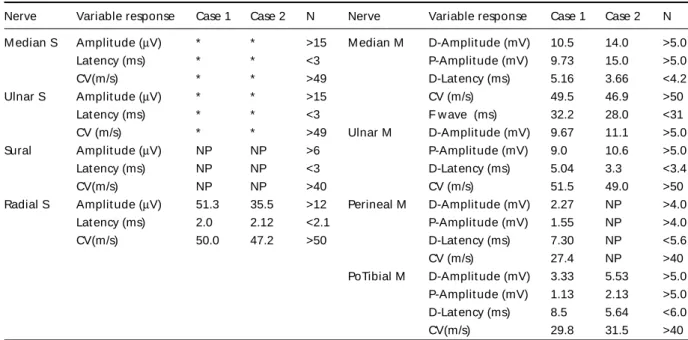
Laboratorial exams (blood count, biochemistry, hepat-ic and renal f unct ion, lipid analysis and coagulat ion t est s) were normal. The biochemical analysis for serum cholesta-nol level, urinary bile alcohol excret ion and chenodeoxy-cholic acid in bile were not done. Cerebrospinal fluid (CSF) w as normal. Elect rocardiogram (ECG) present ed over-load of lef t vent ricle. Elect roencephalogram (EEG) sho-wed mild diffuse cerebral suffering, without irritation activ-it y. Elect rophisiologic st udies show ed sensorimot or demyelinat ing polineuropat hy w it h mild axonal charac-t erischarac-t ics (Table 1). Ophcharac-t halmologic exam evidenced acharac-t ro-phy of retinal pigmented epithelium and peripapillar atro-phy. M agnet ic resonance imaging (M RI) show ed hyper-int ense signal in t he post ero-inf erior segment s of bot h cerebellar hemispheres on T2 WI and ‘FLAIR’ sequences
(Fig 2a). CYP27A1 gene w as sequenced according t o st andard and adequat e t echniques1. It show ed
homozy-gous mut at ion in exon 6 (C1183T) of t he CYP27A1 gene.
Case 2 – A, 40 years old w hit e man, case one´s brot
h-er, which according to the mother’s information, present-ed learning diff icult y since childhood, unable t o read or t o w rit e. He did not f inish t he basic school. At 35 years of age he began w it h progressive diff icult y t o w alk and t w o years after he began to present bilateral swellings of Achil-les t endon. He also present ed chronic diarrhea and w as submitted to a bilateral cataracts surgery in the childhood. Physical examinat ion disclosed no abnormalit ies ex-cept f or bilat eral sw ellings of Achilles t endons (Fig 1b). In the neurological exam he presented mild mental retar-dat ion, moderat e disart hria, visual handicap and f acial palsy w it hout ot her abnormalit ies in cranial nerves. The muscle st rengt h w as V in SL and IV+ in IL (M RC rat ing). Ot her f indings w ere spast icit y in IL w it h preserved t ro-phism, increased deep t endon ref lexes w it h bilat eral Ba-binski and Hof f man signs. Sensibilit y w as preserved. He had w alking dist urbance w it h at axia and spast icit y in IL.
analysis, hepat ic and renal f unct ion) w ere normal. The biochemical analysis f or serum cholest anol level, urina-ry bile alcohol excret ion and chenodeoxycholic acid in bile w ere not done. CSF present ed mild elevat ion of pro-teins (75mg/dl, reference: 35 to 45 mg/dl) with other valu-es in normal limit s. ECG prvalu-esent ed overload of lef t came-ras. EEG present ed w it h mild dif f use cerebral suf f ering, w it hout irrit at ion act ivit y. Elect rophisiologic st udies show ed sensorimot or axonal polineuropat hy w it h mild demyelinat ion (Table 1). He w as submit t ed t o a muscle biopsy w it h normal f resh f rozen sect ion (HE-t richrome, oil red O, PAS, cresyl violet and siryus red) and hist ochem-ical react ions (ATPase, NADH, est erase, myophosphory-lase, acid phosphat ase, alkaline phosphat ase, succinat e dehydrogenase, cyt ochrome-C oxidase and adenylat e deaminase). Opht halmologic exam evidenced incipient cat aract s w it hout surgical indicat ion. M RI w it h hyper-int ense signal in inf erior segment s of bot h cerebellar hemispheres on T2WI and ‘FLAIR’ (Fig 2b). Genet ic analy-sis show ed homozygous mut at ion in exon 6 (C1183T) of t he CYP27A1 gene.
M ot her’s blood w as sequenced and she w as a carrier of t he same exon 6 CYP27A1 gene mut at ion.
The DNA ext ract ion w as made in Curit iba by one au-t hor (SR) and au-t he CYP27A1 analysis w as made in Nijme-gen, The Netherlands (EAS). In both cases we did not start any t reat ment due t o acquisit ion diff icult y of medicat ion and loss of f ollow -up.
DISCUSSION
CTX w as f irst described in t w o cousins, w it h t he presence of cerebellar and pyramidal signs,
pa-lat e myoclonus, cognit ive disabilit y, cat aract s and t endon xant homas, apud Bert ini2. The main
clin-ical charact erist ics are cat aract s and premat ure at herosclerosis, t endon xant homas w it h pref eren-t ial locaeren-t ion f or eren-t he Achilles eren-t endon and neurolog-ical manif est at ions as pyramidal signs, cerebellar at axia, seizures, cognit ive disabilit y, dement ia and peripheral neuropat hy1,3,4. The f indings observed
in our pat ient s w ere cat aract s, t endon xant homas and neurological manif est at ions, such as cerebel-lar at axia, pyramidal signs, cognit ive disabilit y and peripheral neuropat hy suggest ing CTX diagnosis. The combinat ion of chronic diarrhea and bilat eral cat aract s in t he childhood, described in t he case 2, w as pat hognomonic f or t he disease5. Besides cat
a-ract s, ot her ret inal abnormalit ies have been descri-bed in t hese pat ient s as paleness of t he opt ical disk and senile ret inal degenerat ion, as w e could see in t he case 16.
Behavioral dist urbance as depressed or dist im-ic humor, irrit abilit y, appet it e reduct ion, insomnia and fatigue can be present1,3,4. Heart complications,
as lipomatosis hipertrophy with atrial sept thickness and lung alt erat ions and ost eoporosis w ere descri-bed, none w as f ound in our pat ient s7-9.
Other manifestations have been described, such as chronic myelopat hy and spast ic paraplegy in as-sociat ion w it h f ront al lobe dement ia. The diff eren-t ial diagnosis museren-t be done in paeren-t ieneren-t s w ieren-t h early-onset parkinsonism-plus, t hat usually is associat ed Table 1. Nerve conduct ion st udy.
Nerve Variable response Case 1 Case 2 N Nerve Variable response Case 1 Case 2 N
M edian S Amplit ude (µV) * * >15 M edian M D-Amplit ude (mV) 10.5 14.0 >5.0
Lat ency (ms) * * <3 P-Amplit ude (mV) 9.73 15.0 >5.0
CV(m/s) * * >49 D-Lat ency (ms) 5.16 3.66 <4.2
Ulnar S Amplit ude (µV) * * >15 CV (m/s) 49.5 46.9 >50
Lat ency (ms) * * <3 F w ave (ms) 32.2 28.0 <31
CV (m/s) * * >49 Ulnar M D-Amplit ude (mV) 9.67 11.1 >5.0
Sural Amplit ude (µV) NP NP >6 P-Amplit ude (mV) 9.0 10.6 >5.0
Lat ency (ms) NP NP <3 D-Lat ency (ms) 5.04 3.3 <3.4
CV(m/s) NP NP >40 CV (m/s) 51.5 49.0 >50
Radial S Amplit ude (µV) 51.3 35.5 >12 Perineal M D-Amplit ude (mV) 2.27 NP >4.0
Lat ency (ms) 2.0 2.12 <2.1 P-Amplit ude (mV) 1.55 NP >4.0
CV(m/s) 50.0 47.2 >50 D-Lat ency (ms) 7.30 NP <5.6
CV (m/s) 27.4 NP >40
PoTibial M D-Amplit ude (mV) 3.33 5.53 >5.0 P-Amplit ude (mV) 1.13 2.13 >5.0 D-Lat ency (ms) 8.5 5.64 <6.0
CV(m/s) 29.8 31.5 >40
w it h w alking dist urbances, pyramidal signs, cogni-t ive abnormalicogni-t ies and parcogni-t ial L-dopa response10-12.
The diagnosis can be conf irmed by biochemi-cal exams, which demonstrate increase of serum cho-lest anol level and urinary bile alcohol excret ion associat ed w it h reduct ion of t he chenodeoxycholic acid in bile13. Because technical difficulties these
tes-t s w ere notes-t done in our pates-t ientes-t s.
M olecular genet ic analysis, accomplished in our pat ient s, is obt ained by t he screening of CYP27A1 gene locat ed in chromosome 2q 23. The genomic structure of CYP27A1 contains nine exons and eight int rons w it h 18,6 kb of DNA. The mat ure enzyme consists of 498 amino acids and it is expressed in CNS, liver, lungs, duodenum and endot helial cells. Unt il now, more than 45 mutations were described in t he CYP27A1 gene, usually aff ect ing heme or adreno-doxin relat ed domains bet w een exons 6 and 91,13-15.
The pat ient s described here present ed a homozy-gous CYP27A1 gene mut at ion in exon 6 (1183 C>T) t hat w as already described in ot her populat ions1,16.
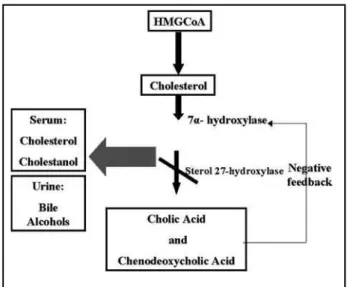
CYP27A1 enzyme (st erol 27-hydroxylase) w it h t w o prot ein cof act ors, adrenodoxin and adrenodo-xin reduct ase, hydroxylat es a variet y of st erols at t he C-27 posit ion producing t he primary bile acids (cholic and chenodeoxycholic acid)14. In normal
in-dividuals, t he primary bile acid inhibit s t he rat e-limit ing st ep in t heir product ion, t he enzyme 7 µ
-hydroxylase (negat ive f eedback mechanism). The gene mutation causes deficiency of the enzyme
ste-rol 27-hydroxylase, leading t o reduced synt hesis of cholic acid and almost no chenodeoxycholic acid. Due t o t he absence of t he negat ive f eedback mecha-nism, t he 7 ?-hydroxylase act ivit y is increased, pro-ducing excessive cholest erol and cholest anol and accumulat ing t hem in many t issues w it h increased urine excret ion of t he bile alcohols2,17. The met
abo-lic pat hw ay can be seen in t he Figure 3.
The electrophisiologic findings from CTX patients usually show an axonal degeneration, but they com-monly are described as a mixed neuropat hy, as ob-served in our cases1. M uscle biopsy is uncharact
eris-t ic4.Xant homat ous lesions, lipid cryst al clef t s and
macrophage clust ers t hat are usually seen in t he CNS, w ere not f ound in t he muscle biopsies4. M RI
usually demonst rat e hyperint ense signal in T2 and FLAIR locat ed pref erent ially in perivent ricular area, basal ganglia and dent at e nuclei of t he cerebel-lum, besides variable degrees of cerebral and cere-bellar at rophy (Fig 2A)18-21. Brain spect roscopy
de-monst rat es signif icant decreases of NAA (N-acet y-laspart at e) and increases of lact at e M RI signals. These result s suggest s a brain accumulat ion of me-tabolic neurotoxins and deficiency of the mitochon-dria metabolism secondary to the high levels of cho-lest anol and bile alcohols21,22.
The mechanism f or neurological manif est at ions is not know n. A hypot hesis suggest s t hat t his hap-pens from an increase neuronal apoptosis caused by t he cholest anol overload. The presence of apolipo-prot ein B in CSF indicat es penet rat ion of low -den-sit y lipoprot ein part icles f rom plasma t hrough t he blood-brain barrier. These lipoprot ein part icles may carry cholestanol as well as cholesterol13.
Chenodeo-xycholic acid replacement t herapy is usually asso-ciat ed w it h improvement in t he clinical sympt oms, normalization cholesterol synthesis and reestablish-ment of select ive permeabilit y of blood-brain bar-rier w it h normalized CSF apolipoprot ein and st erol concent rat ions23. The absence of neurological
dys-f unct ion in ot her lipid disorders (dys-f amilial hypercho-lest erolemia or sit ost erolemia) f urt her support t he hypot hesis t hat cholest anol it self impairs brain f unct ion13.
It is important to remember that precocious diag-nosis is fundamental due to the good response with chenodeoxycholic acid t reat ment reducing choles-t anol accumulacholes-t ion and promocholes-t ing lesion regres-sion. The eff ect iveness of st at ins is cont roversial w i-t h i-t he possibilii-t y of w orsening i-t he condii-t ion ow n-ing t o increased low -densit y lipoprot ein upt ake as t he result of augment ed low -densit y lipoprot ein Fig 3. Enzymat ic mechanism in Cerebrot endinous xant homat
recept or act ivit y13. The det ect ion of t he specif ic
mut at ion in each f amily is import ant t o def init ely conf irm t he diagnosis, t o st art t reat ment as earli-er as possible, and to provide accurate genetic coun-seling t o t he pat ient s and t heir f amily.
Acknow ledgem ent s - The aut hors w ish t o t hanks
Dr. Ana C. Bacelar Limeira f rom CETAC - Curit iba f or neu-roimage evaluat ion.
REFERENCES
1. Verrips A, Hoesfloot LH, Steenbergen GCH, et al. Clinical and molecu-lar genetic characteristics of patients with cerebrotendinous xanthomato-sis. Brain 2000;123:908-919.
2. Bertini E, Dionisi-Vici C, Zeviani M. Metabolic and mitochondrial ataxi-as. In Pulst SM (ed). Genetics of movement disorders. San Diego: Elsevier,2003:238.
3. Lee Y, Lin PY, Chiu NM, Chang WN, Wen JK. Cerebrotendinous xantho-matosis with psychiatric disorders: report of three siblings and litera-ture review. Chang Gung Med J 2002;25:334-340.
4. Verrips A, van Engelen BGM, ter Laak H, et al. Cerebrotendinous xan-thomatosis: controversies about nerve and muscle: observations in ten patients. Neuromusc Disord 2000;10:407-414.
5. Cruysberg JRM, Wevers RA, Tolboom JJM. Juvenile cataract associa-ted with chronic diarrhea in pediatric cerebrotendinous xanthomato-sis. Am J Ophthalm 1991;112:606-607.
6. Dotti MT, Rufa A, Federico A. Cerebrotendinous xanthomatosis: hete-rogeneity of clinical phenotype with evidence of previously undescri-bed ophthalmological findings. J Inherit Metab Dis 2001;24:696-706. 7. Kawabata M, Kuriyama M, Mori S, Sakashita I, Osame M. Pulmonary
manifestations in cerebrotendinous xanthomatosis. Intern Med 1998;37:922-926.
8. Dotti MT, Mondillo S, Plewnia K, Agricola E, Federico A. Cerebroten-dinous xanthomatosis: evidence of lipomatous hypertrophy of the atri-al septum. J Neurol 1998;245:723-726.
9. Berginer VM, Shany S, Alkalay D, et al. Osteoporosis and increased bone fractures in cerebrotendinous xanthomatosis. Metabolism 1993;42:69-74.
10. Verrips A, Nijeholt GJ, Barkhof F, et al. Spinal xanthomatosis: a variant of cerebrotendinous xanthomatosis. Brain 1999;122:1589-1595. 11. Sugama S, Kimura A, Chen W, et al. Frontal lobe dementia with
abnor-mal cholesterol metabolism and heterozygous mutation in sterol 27-hydroxylase gene (CYP27). J Inherit Metab Dis 2001;24:379-392. 12. Grandas F, Anaya F. Early-onset parkinsonism in cerebrotendineous
xan-thomatosis. Mov Disord 2002;17:1396-1397
13. Moghadasian MH, Salen G, Frohlich JJ, Scudamore CH. Cerebrotendi-nous xanthomatosis: a rare disease with diverse manifestations: arch Neurol 2002;59:527-529.
14. Cali JJ, Russell DW. Characterization of the human sterol 27-hydroxyla-se: a mitochondrial P-450 that catalyzes multiple oxidation reactions in bile acid biosynthesis. J Biol Chem 1991;266:7774-7778.
15. Lamon-Fava S, Schaefer EJ, Garuti R, Salen G, Calandra S. Two novel mutations in the sterol 27-hydroxylase gene causing cerebrotendinous xanthomatosis. Clin Genet 2002;61:185-191.
16. Cali JJ, Hsieh CL, Francke U, Russell DW. Mutations in the bile acid biosynthetic enzyme sterol 27-hydroxylase underlie cerebrotendinous xanthomatosis. J Biol Chem 1991;266:7779-7783.
17. Oftebro H, Björkhem I, Stormer FC, Pedersen JL. Cerebrotendinous xan-thomatosis: defective liver mitochondrial hydroxylation of chemode-oxycholic acid precursors. J Lipid Res 1981;22:632-640.
18. Dotti MT, Federico A, Signorini E, et al. Cerebrotendinous xanthomato-sis (van Bogaert-Scherer-Epstein disease): CT and MR findings. AJNR 1994;15:1721-1726.
19. Hokezu Y, Kuriyama M, Kubota R, Nakagawa M, Fujiyama J, Osame M. Cerebrotendinous xanthomatosis: cranial CT and MRI studies in eight patients. Neuroradiology 1992;34:308-312.
20. Vanrietvelde F, Lemmerling M, Mespreuve M, Crevits L, De Reuck J, Kunnen M. MRI of the brain in cerebrotendinous xanthomatosis (van Bogaert-Scherer-Epstein disease). Eur Radiol 2000;10:576-578. 21. De Stefano N, Dotti M, Mortilla M, Frederico A. Magnetic resonance
imaging and spectroscopic changes in brains of patients with cerebroten-dinous xanthomatosis. Brain 2000;124:121-131.
22. Soffer D, Benharroch D, Berginer V. The neuropathology of cerebrotendi-nous xanthomatosis revisited: a case report and review of the literatu-re. Acta Neuropathol (Berl) 1995;90:213-220.