J. Braz. Chem. Soc. vol.17 número7
Texto
Imagem

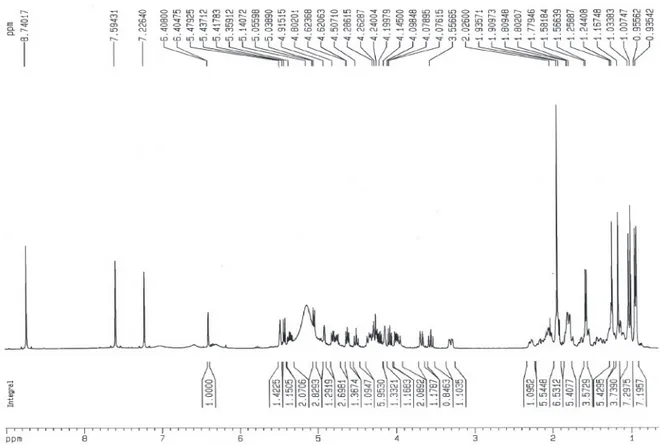
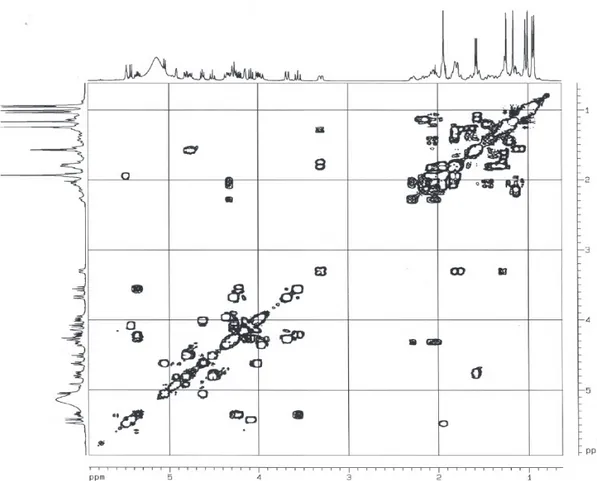
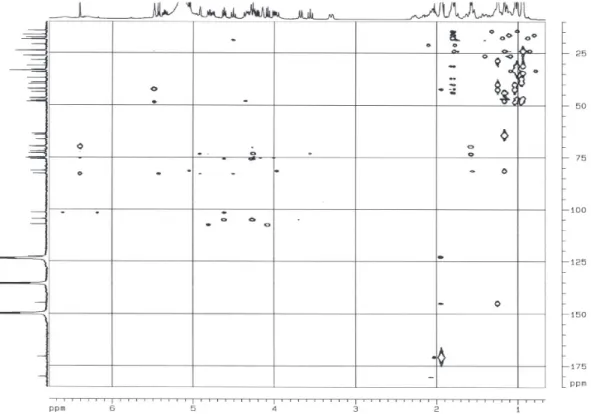
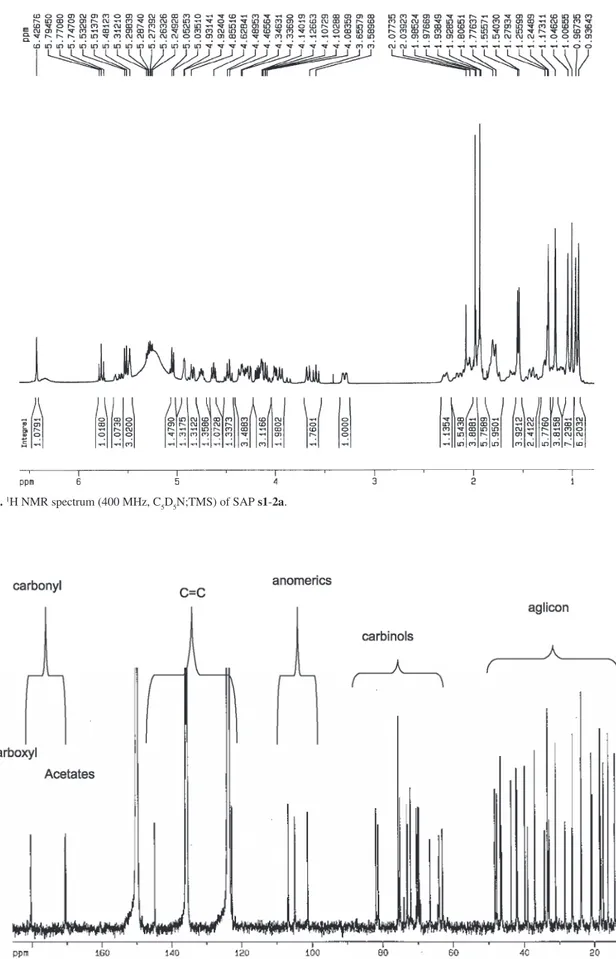
Documentos relacionados
Brazilian tropical Latosol soil samples were thermally analyzed associating thermogravimetric (TG) and differential scanning calorimetric (DSC) techniques under synthetic air or
In this study a spectrophotometric method for the determination of the trace amounts of dopamine, methyldopa and levodopa has been introduced based on their oxidation reaction
Compound 2 was screened in vitro in seven human cancer cell lines, MCF7 and EVSAT (mammary cancers), WiDr (colon cancer), IGROV (ovarian cancer), M19 (melanoma), MEL A498
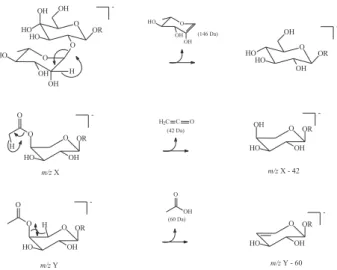
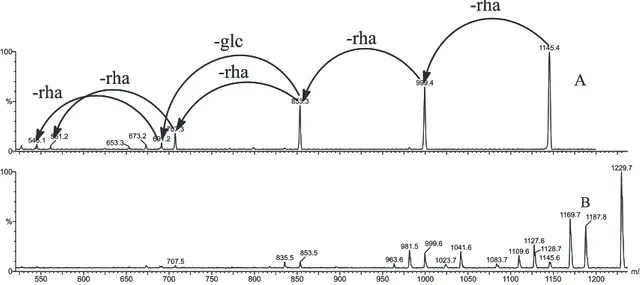
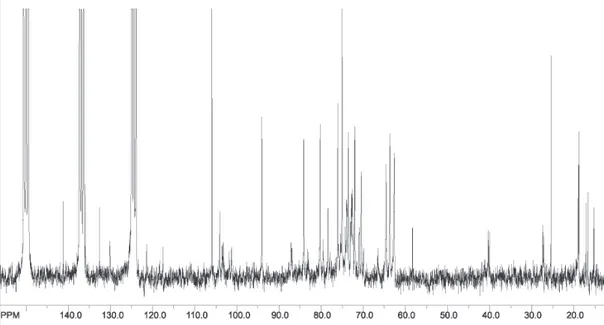
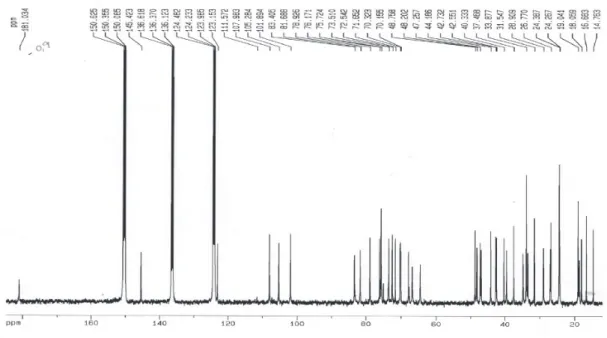
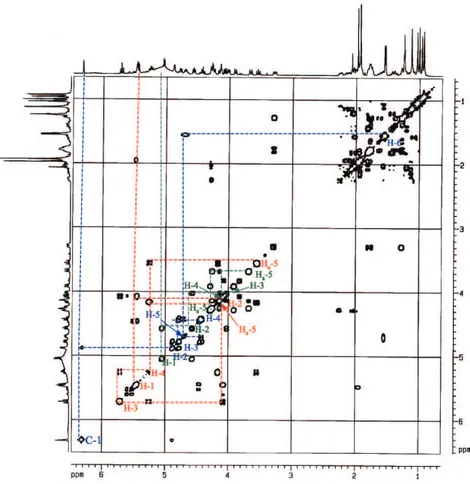
The phytochemical study of the stem bark of Aspidosperma nitidum led to the isolation of a new type of indole alkaloid with a 1,2,9-triazabicyclo[7.2.1] system, which has been
The methanolysis of soybean oil carried out in the presence of solid 3 achieved a conversion yield of 30% after 4 h using the same experimental conditions as used when solids 1 and
In Table 3, the major interfering ions (just a gross relative comparison), dynamic linear range, the detection limit and response time of the proposed electrode are compared with
Curso de Pós-Graduação em Química Orgânica, Departamento de Química Orgânica e Inorgânica, Universidade Federal do Ceará, CP 12.200, 60455-760 Fortaleza-CE, Brazil. Setor de
Thus, at first, FPD was used as a neutral carrier to prepare PVC-based membrane electrodes for a variety of trivalent transition metal ions, including lanthanum, cerium,
