UFOP - CETEC - UEMG
REDEMAT
REDE TEMÁTICA EM ENGENHARIA DE MATERIAIS
UFOP – CETEC – UEMG
Tese de Doutorado
“
Lixiviação de bornita, calcopirita e dos produtos da
sulfidização desta última em meio híbrido
sulfato-cloreto
”
Autora: Tácia Costa Veloso
Orientador: Prof. DSc. Versiane Albis Leão
UFOP - CETEC - UEMG
REDEMAT
R
EDET
EMÁTICA EME
NGENHARIA DEM
ATERIAISUFOP – CETEC – UEMG
Tácia Costa Veloso
“
Lixiviação de bornita, calcopirita e dos produtos da sulfidização desta
última em meio híbrido sulfato-cloreto
”
Tese de doutorado apresentada ao Programa de Pós-Graduação em Engenharia de Materiais da REDEMAT, como parte integrante dos requisitos para obtenção do título de Doutora em Engenharia de Materiais.
Área de concentração: Processos de fabricação
Orientador: Prof. DSc. Versiane Albis Leão
Catalogação: www.sisbin.ufop.br V443l Veloso, Tácia Costa.
Lixiviação de bornita, calcopirita e dos produtos da sulfidização desta última em meio híbrido sulfato-cloreto [manuscrito] / Tácia Costa Veloso. - 2016. 121f.: il.: color; grafs; tabs.
Orientador: Prof. Dr. Versiane Albis Leão.
Dissertação (Mestrado) - Universidade Federal de Ouro Preto. Escola de Minas. Engenharia Metalúrgica e de Materiais. Rede Temática em Engenharia de Materiais.
1. Minérios de cobre. 2. Bornita. 3. Teoria cinética da matéria. 4.
Calcopirita. 5. Lixiviação. I. Leão, Versiane Albis. II. Universidade Federal de Ouro Preto. III. Titulo.
ii
iii
SUMÁRIO
RESUMO ... 14
ABSTRACT ... 16
1 CAPÍTULO 1 - INTRODUÇÃO ... 18
1.1 Contextualização e organização da tese ... 18
1.2 Objetivos ... 21
2 CAPÍTULO 2 - REFERENCIAL TEÓRICO ... 23
2.1 Parâmetros que influenciam na lixiviação de calcopirita ... 23
2.2 Parâmetros que influenciam na lixiviação da bornita ... 26
2.3 Mecanismos de lixiviação da bornita ... 28
2.4 Cinética de lixiviação da calcopirita e bornita ... 32
2.5 A reação entre calcopirita e enxofre ... 41
2.6 Referências Bibliográficas ... 46
3 CAPÍTULO 3 – CINÉTICA DE LIXIVIAÇÃO DA CALCOPIRITA ... 52
3.1 Introduction ... 53
3.2 The effect of PSD on the SCM ... 56
3.3 Experimental Details ... 58
3.3.1 Sample Preparation ... 58
3.3.2 Leaching tests ... 59
3.4 Results and discussion ... 60
3.4.1 Effect of the type of leaching agent on chalcopyrite leaching ... 60
3.4.2 Effect of temperature on the leaching of chalcopyrite. ... 63
3.4.3 Kinetics of chalcopyrite leaching ... 65
3.5 Conclusions ... 71
3.6 Acknowledgements ... 72
3.7 References ... 72
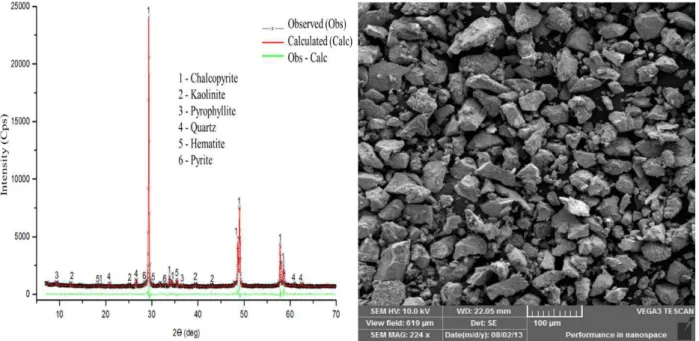
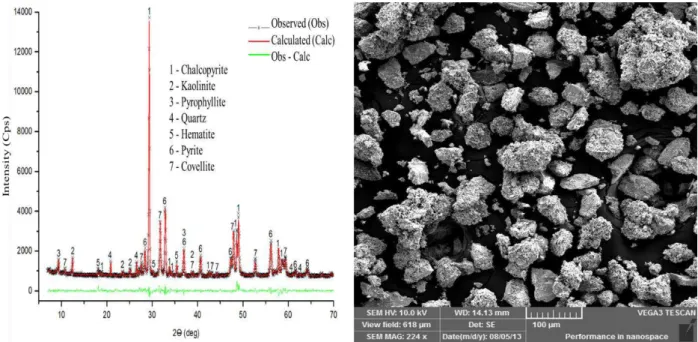
4 CAPÍTULO 4 – TRANSFORMAÇÃO DA CALCOPIRITA EM OUTRO SULFETO DE COBRE ... 77
4.1 Introduction ... 78
4.2 Materials and methods... 80
4.2.1 Sample and reaction product characterization ... 80
iv
4.2.3 Leaching tests ... 81
4.3 Results and discussion ... 82
4.3.1 Sulfurization of chalcopyrite ... 82
4.3.2 Leaching of sulfurization products ... 89
4.4 Conclusions ... 94
4.5 Acknowledgements ... 94
4.6 References ... 95
5 CAPÍTULO 5 – CINÉTICA DE LIXIVIAÇÃO DA BORNITA ... 99
5.1 Introduction ... 100
5.2 Mechanism of bornite leaching ... 101
5.3 Materials and methods... 103
5.3.1 Sample and reaction product characterization ... 103
5.3.2 Leaching tests ... 104
5.4 Results and discussion ... 105
5.4.1 Characterization of the concentrate and the leaching residue ... 105
5.4.2 Main parameters affecting copper extraction from bornite... 110
5.4.3 Bornite leaching kinetics ... 113
5.5 Conclusions ... 117
5.6 Acknowledgements ... 117
5.7 References ... 117
6 CAPÍTULO 6 - CONSIDERAÇÕES FINAIS ... 121
6.1 Principais Conclusões e Contribuições ... 121
v
AGRADECIMENTOS
Na realização do presente trabalho, contei com o apoio direto ou indireto de múltiplas pessoas e instituições às quais estou profundamente grata. Correndo o risco de injustamente não mencionar alguém, deixo meus agradecimentos:
Agradeço primeiramente a Deus, que me guia e me dá forças para prosseguir. E desta vez foi preciso de muita.
Ao Professor Versiane Albis Leão, por ter me orientado desde o mestrado, proporcionado valiosas discussões científicas, oportunidades de realização de trabalhos que vão além deste, infraestrutura e tudo o que fosse necessário para o desenvolvimento da pesquisa.
Ao Professor Carlos Antônio da Silva, pela ajuda com o aparato experimental, concessão de espaço em seu laboratório e acima de tudo por ser meu maior exemplo de professor.
A toda equipe do laboratório de Bio&Hidrometalurgia, Damaris Guimarães, Flávia Donária, Flávio Martins, Flávio Luciano, Isabel Braga, Larissa Melgaço, Marcio Salgado, Michael Marques, Renata Castro, Sergio e Sueli Bertolino, pelas experiências compartilhadas.
Aos bolsistas de iniciação científica, Johne Peixoto por ser brilhante e tão responsável; Gabriela Maciel pela dedicação e amizade; Lorena Teixeira e Carlos Henrique pela ajuda na execução dos ensaios.
Aos técnicos Graciliano Dimas Francisco e Paulo Sérgio Moreira pela preparação das amostras para análises microscópicas.
vi Ao Laboratório de Caracterização do Departamento de Engenharia de Materiais (CEFET/MG- Campus I), especialmente ao Paulo Renato Perdigão de Paiva, pelas análises de DRX e quantificação das fases minerais.
À Professora Rosa Malena Lima e ao Laboratório de Tratamento de Minérios DEMIN-UFOP, pelas análises de Distribuição Granulométrica.
À Professora Gláucia Nascimento Queiroga, ao Marco Paulo de Castro e ao Laboratório de Microanálises DEGEO/UFOP, pelas análises de microssonda eletrônica.
Aos colegas do Departamento de Engenharia Metalúrgica e de Materiais, que incentivaram a realização deste trabalho.
Ao Professor Edison Tazava, por gentilmente ter fornecido amostras de minérios.
A Capes, pela concessão da bolsa de doutorado, sem a qual este trabalho não poderia ser realizado;
Às moradoras e ex-alunas da República Indignação, pela amizade e carinho de sempre. Vocês são minha família em Ouro Preto. Obrigada pelos bons momentos de descontração, os almoços de domingo e inúmeras risadas. Com certeza estes momentos contribuíram para que esta caminhada fosse mais leve.
Aos amigos de Três Pontas, pela constante acolhida. Especialmente a Isabela Garcia, por ser minha irmã de coração, a Giselle Tardiloli, por ser minha dupla desde os tempos da pré-escola, Eloá Teixeira, pelas inúmeras experiências e confidências compartilhadas, Georgia Duque, por sempre se fazer presente. Ainda neste time agradeço a Fernanda Ferraz, Alessandra Oliveira, Danielle Tardioli, Andrea Brito, Valéria Brito e Laiz Figueiredo, apesar da distância, levo vocês no coração.
À minha querida Chica, por ser minha mais fiel companheira.
viii
LISTA DE FIGURAS
Figura 2. 1 Variação da pressão de vapor do enxofre em função da temperatura. ... 42 Figura 2. 2 Curvas de sulfidização de alguns sulfetos em função da temperatura. ... 43
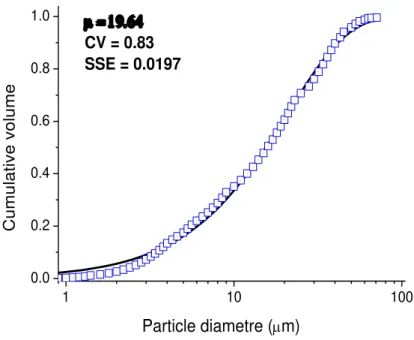
Figure 3. 1 Particle size distribution of the chalcopyrite sample under study. ... 59 Figure 3. 2 Effect of concentration of NaCl on chalcopyrite dissolution. 1.0 mol/L H2SO4,
1.0 mol/L Fe3+ and 80 °C. PSD following the Rosin-Rammler distribution (µ = 19.6 µm and CV
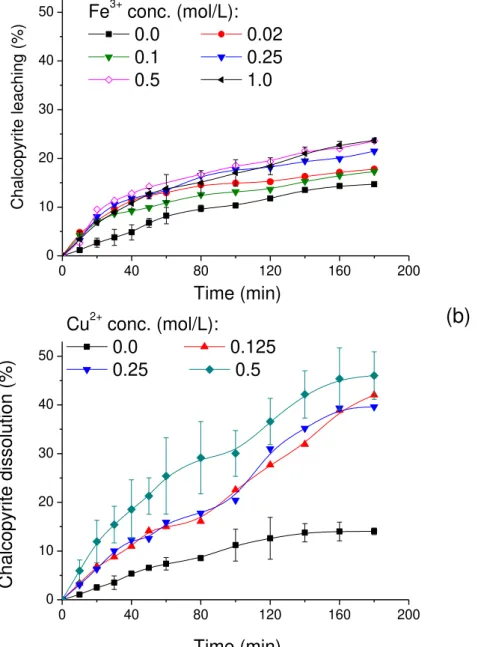
= 0.83). ... 61 Figure 3. 3 Effect of Fe3+ (a) and Cu2+ (b) concentrations on chalcopyrite dissolution at 1.0 mol/L
H2SO4, 2.0 mol/L NaCl and 80 °C. PSD following the Rosin-Rammler distribution (µ = 19.6 µm
and CV = 0.83) ... 62 Figure 3. 4 Effect of temperature on chalcopyrite dissolution: (a) 1.0 mol/L Fe3+ and (b) 0.5 mol/L
Cu2+. 1.0 mol/L H
2SO4, 2.0 mol/L NaCl. The PSD followed the Rosin-Rammler distribution (µ =
19.6 µm and CV = 0.83). ... 64 Figure 3. 5 SEM-EDS analysis of the leaching residue (69.6% dissolution) produced during chalcopyrite leaching in chloride media. Experimental conditions: 0.5mol/L Cu2+, 2.0mol/L NaCl, 1.0mol/L H2SO4, 90°C. ... 66
Figure 3. 6 Fitting of experimental data to the PSD-incorporated SCM model (chemical control) at different temperatures. Experimental conditions: 1.0 mol/L Fe3+ (a) and 0.5 mol/L Cu2+ (b) in
the presence of 1.0 mol/L H2SO4 and 2.0 mol/L NaCl. The PSD followed the Rosin-Rammler
distribution (µ = 19.6 µm and CV = 0.83). ... 69 Figure 3. 7 Arrhenius plot for the Fe3+ -Cl- and Cu2+ -Cl- leaching of chalcopyrite. Experimental
conditions: 1.0 mol/L Fe3+ or 0.5 mol/L Cu2+ in the presence of 1.0 mol/L H
2SO4 and 2.0 mol/L
ix Figure 4. 1 X-ray difractogram after applying Rietveld refining method and SEM surface appearance; activation at 573 K (300 °C). ... 83 Figure 4. 2 X-ray difractogram after applying Rietveld refining method and SEM surface appearance; activation at 623 K (350 °C). ... 84 Figure 4. 3 X-ray difractogram after applying Rietveld refining method and SEM surface appearance; activation at 673 K (400 °C). ... 84 Figure 4. 4 X-ray difractogram after applying Rietveld refining method and SEM surface appearance; activation at 723 K (450 °C). ... 85 Figure 4. 5 An overview of the surface appearance of the original chalcopyrite and synthetic bornite; SEM (1000x). Points 1, 2 and 3 are chalcopyrite; 4 and 5 silicates; 6 and 7 bornite; 8 and 9 pyrite. ... 88 Figure 4. 6 Chemical and superficial appearance of original chalcopyrite concentrate (a) and products of sulfurization (b) (synthetic bornite); analysis by SEM and EDS (5000x). ... 89 Figure 4. 7 Leaching behavior of original chalcopyrite and activation products. Synthetic Bornite (chalcopyrite reaction product after sulfurization at 723 K (450 °C) during 60min) Synthetic Covellite (chalcopyrite reaction product after sulfurization at 623 K (350°C) during 120 min). Leaching conditions:0.5 mol/L de Fe3+, 2.0 mol/L de NaCl, 1.0 mol/L de H
2SO4, at 353K (80°C). ... 90 Figure 4. 8 Copper extraction from synthetic bornite as a function of initial ferric ion concentration. Leaching conditions: 0 to 0.5 mol Fe3+/L, 2.0 mol/L NaCl, 1.0 mol/L H
2SO4, at 353K (80°C). 91 Figure 4. 9 Effect of temperature on the dissolution rate of synthetic bornite (a) and natural bornite (b). Leaching conditions:0.5 mol/L Fe3+, 2.0 mol/L NaCl and 1.0 mol/L H
2SO4. ... 92 Figure 4. 10 Superficial appearance of bornite concentrate (a) and products of sulfurization (b) (synthetic bornite); analysis by SEM (4000x). ... 94
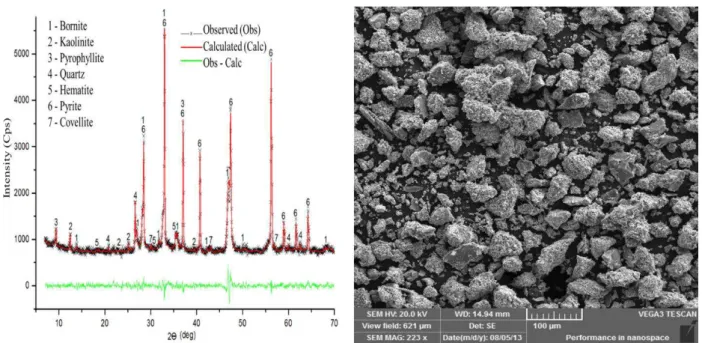
x Figure 5. 2 X-ray difractogram of the leaching residue (65.2% dissolution) produced during bornite leaching. Experimental conditions: 0.5mol /L Cu2+, 2.0 mol/L NaCl, 1.0 mol/L H
2SO4, at 353K (80°C). ... 106 Figure 5. 3 SEM-EDS analysis of the leaching residue (65.2% dissolution) produced during bornite leaching. Experimental conditions: 0.5 mol /L Cu2+, 2.0 mol/L NaCl, 1.0 mol/L H
2SO4, at 80°C
(1000x). ... 107 Figure 5. 4 Electron microprobe chemical compositions of initial bornite. ... 108 Figure 5. 5 Electron microprobe chemical compositions of the leaching residue (65.2% dissolution) produced during bornite leaching. . Experimental conditions: 0.5 mol/L Cu2+,
2.0 mol/L NaCl, 1.0 mol/L H2SO4, at 80°C (1000x). ... 108
Figure 5. 6 SEM-EDS analysis of initial bornite concentrate (4000x). ... 109 Figure 5. 7 SEM-EDS analysis of the leaching residue (65.2% dissolution) produced during bornite leaching. Experimental conditions: 0.5 mol/L Cu2+, 2.0 mol/L NaCl, 1.0 mol/L H
2SO4, at 80°C
(4000x). ... 109 Figure 5. 8 Effect of particle size on bornite dissolution. Leaching conditions: 0.5 mol/L Cu2+,
2.0 mol/L NaCl, 1.0 mol/L H2SO4, at 353K (80 °C). ... 110
Figure 5. 9 Effect of Cu2+ concentrations on bornite dissolution at 1.0 mol/L H
2SO4, 2.0 mol/L
NaCl, 353K (80°C) and particle size -53 μm +45μm. ... 112 Figure 5. 10 Effect of temperature on bornite dissolution. Leaching conditions: 0.5 mol/L Cu2+,
1.0 mol/L H2SO4, 2.0 mol/L NaCl and particle size -53 μm +45μm. ... 113
Figure 5. 11 Fitting of experimental data to the grain model (chemical control) at different temperatures. Leaching conditions: 0.5 mol/L Cu2+, 1.0 mol/L H
2SO4, 2.0 mol/L NaCl and particle size -53 μm +45μm. ... 116 Figure 5. 12 Arrhenius plot for bornite leaching. Leaching conditions: 0.5 mol/L Cu2+, 1.0 mol/L
xi
LISTA DE TABELAS
Tabela II. 1: Distribuição de tamanho de partículas comumente empregada ... 37
Table III. 1: Values of Krn at different temperatures achieved during chalcopyrite leaching by either Fe3+/Fe2+ or Cu2+/Cu+ couples. Experimental conditions: 1.0 mol/L Fe3+ or 0.5 mol/L Cu2+
in the presence of 1.0 mol/L H2SO4 and 2.0 mol/L NaCl. The PSD followed the Rosin-Rammler
distribution (µ = 19.6 µm and CV = 0.83). ... 70
Table IV. 1: Relative amount of sulfide phases produced by thermal activation; reaction time of 60minutes). ... 85 Table IV. 2: Relative amount of sulfide phase obtained from thermal activation at 635 K (350 °C) as a function of reaction time. ... 87
xii
LISTA DE SÍMBOLOS
SCM shrinking core model ou modelo do núcleo não reagido
PSD particle size distribution ou distribuição do tamanho de partícula
XB fraction of B in solid that is converted to products ou fração de B convertida em produtos
t time ou tempo
km rate constant film diffusion control ou constante de velocidade para controle por difusão na
camada limite
b mols de B consumido por mols de A reagido;
kg coeficiente de transferência de massa de A na camada limite (m/s)
CAb concentração de A no seio do líquido (mol/m3) ρ densidade molar de B (mol/m3)
r raio da partícula (m)
kd rate constant for ash layer diffusion control constante de velocidade para controle por difusão
na camada de cinzas
De coeficiente de difusão efetiva de A através da camada de cinzas (m2/s)
kr rate constant for chemical control ou constante de velocidade para controle químico
k1 constante de velocidade da reação química (m(3n-2)/mol(n-1).s)
n ordem de reação em relação a A k constante de velocidade
k0 fator pré-exponencial da equação de Arrhenius
Ea energia de ativação
R constante real dos gases T temperatura
µ mean ou tamanho médio de partículas CV covariance ou coeficiente de variação
CNTP condições padrão de pressão e temperatura ΔG° energia livre padrão de Gibbs
α atividade das espécies
CESL Comico Engineering Services Limited
xiii EDS energy dispersive spectroscopy
PPS pseudo-steady state D diameter
krn krD
kdn kdD2
kmn kmD
FU fraction of unreacted particles
Dmax the size density of the largest particle in the system
p(D) particle size density function based on mass of particles
m and l positive constants
𝛤 gamma function
SSE sum of squared errors
𝑋𝐵𝑒𝑥𝑝 fraction reacted experimental values 𝑋𝐵𝑝𝑟𝑒𝑑 fraction reacted predicted values EMPA electron microprobe analyses X degree of conversion
kR apparent chemical reaction rate constant, according to the grain model
CAS cupric ions concentration at the surface of grains
B bornite molar density
rg initial grain radius
kD apparent mass transfer coefficient, according to the grain model
CA0 cupric ions concentration in the bulk
r0 initial particle radius
Deff effective diffusion coefficient
14
RESUMO
Apesar das intensivas pesquisas realizadas nos últimos cinquentas anos, a lixiviação da calcopirita ainda é um desafio tanto do ponto de vista acadêmico quanto industrial. Na presente tese investigou-se a lixiviação da calcopirita de duas formas diferentes: (i) dissolução da calcopirita em meio contento sulfato e cloreto (este meio foi escolhido devido ao efeito catalítico dos íons cloretos na lixiviação e considerando o aumento da utilização de água salina em processos hidrometalúrgicos); (ii) sulfidização da calcopirita com enxofre elementar, convertendo-a em bornita e covelita, que são sulfetos de cobre que apresentam cinética de lixiviação mais favorável. A investigação foi concluída com uma análise cinética da lixiviação da bornita e um mecanismo para a dissolução desta fase com íons cúpricos foi proposto. Primeiramente, uma extensão do SCM (shrinking core model) que leva em consideração a PSD (particle size distribution) da amostra foi
empregada para determinar os parâmetros cinéticos da lixiviação da calcopirita. Especificamente, os efeitos da temperatura (70 °C a 90 °C) e concentração dos reagentes Cl-, Fe3+ e Cu2+ na cinética
de lixiviação foram avaliados. Os resultados mostraram que a calcopirita foi mais rapidamente lixiviada com íons Cu2+ (maior constante de velocidade) do que com íons férricos, porém os
valores de energia de ativação aparente em ambos os sistemas de lixiviação foram semelhantes, ou seja, 66,6 kJ/mol com 0,5 mol/L de Cu2+ e 66,8 kJ/mol com 1,0 mol/L Fe3+. Subsequentemente,
a sulfidização da calcopirita objetivando produzir bornita e covelita foi estudada. O concentrado de cobre foi reagido com enxofre elementar em um forno tubular e no intervalo de 300 °C a 450 °C, seguido de lixiviação atmosférica dos produtos de reação por Fe3+. As fases minerais foram
quantificadas utilizando o método de Rietveld, e foi observado que a calcopirita foi convertida em covelita (41%) e pirita (34%), nos ensaios conduzidos em temperaturas abaixo de 400 oC. Quando
a temperatura estava acima de 400 oC, os produtos de reação foram bornita (45%) e pirita (31%).
15 bornita de maneira mais significativa foi a temperatura, resultando em um aumento de 11,3% a 30°C para 100% a 90°C. Devido ao aparecimento das trincas e à presença dos subcristalitos durante a lixiviação, o modelo cinético utilizado para descrever a dissolução da bornita com íons cúpricos foi o modelo do grão. Os dados experimentais apresentaram um bom ajuste ao controle químico e energia de ativação aparente obtida foi de 65.27 kJ/mol. Os interiores dos grãos apresentaram a composição química do intermediário Cu3FeS4, determinada por microssonda
eletrônica e em função deste intermediário um mecanismo para dissolução foi proposto.
16
ABSTRACT
Despite extensive research in the last fifty years, chalcopyrite leaching still remains as important challenge to both industry and academy. The current thesis addressed chalcopyrite leaching through two different approaches: (i) leaching in mixed sulphate-chloride systems since chloride is a well know catalyst of its dissolution and also due to an increasing utilization of saline waters in both leaching and bioleaching of sulphide ores; (ii) chalcopyrite sulfurization by elemental sulphur to bornite and covellite, both presenting fast leaching kinetics. The investigation was concluded with a kinetics analysis of bornite leaching in chloride systems and a proposed mechanism for its leaching by cupric ions. Firstly, an extension of the SCM (particle size distribution) was successfully applied to the leaching of a chalcopyrite sample with a broad PSD (particle size distribution) in a mixed chloride-sulphate solution. Specifically, the effects of temperature (70 °C to 90 °C) and reagent (Fe3+, Cu2+ and Cl-) concentrations on the leaching
kinetics were determined. The results showed that chalcopyrite leaching was faster with Cu2+
(larger rate constant) than with Fe3+, but the apparent activation energy was similar in both cases,
with 66.6 kJ/mol for 0.5 mol/L of Cu2+ and 66.8 kJ/mol with 1.0 mol/L Fe3+. Subsequently,
chalcopyrite sulfurization aiming to produce bornite and covelite was investigated. A copper concentrate was reacted with elemental sulfur in a tubular furnace at temperatures ranging from 300 °C to 450 °C, followed by atmospheric leaching in an Fe3+-bearing solution. The mineral
17 was selected to determine leaching kinetics parameters. Bornite leaching by cupric ions has been shown to be chemically controlled with an apparent activation energy of 65.27 kJ/mol. In addition, microprobe analysis revealed the formation of Cu3FeS4 as an intermediary product, and based on
this, a mechanism for the Cu2+ leaching of bornite was proposed.
18
1 CAPÍTULO 1 - INTRODUÇÃO
1.1 Contextualização e organização da tese
A calcopirita (CuFeS2) é o mineral de cobre mais abundante em termos de reservas mundiais,
representando cerca de 70% dos depósitos conhecidos, e, por isso, é a mais importante fonte de cobre para a produção do metal (KAPLUN et al., 2011). Normalmente, a produção do cobre a
partir de concentrados contendo este mineral ocorre através da rota pirometalúrgica, ou seja, envolvendo as etapas de fusão à matte, conversão, refino ao fogo e eletrorrefino (MUNOZ et al.,
1979). Entretanto, as reservas mundiais de cobre vêm apresentando minérios com teores de cobre cada vez mais baixos e mineralogias complexas, fato que dificulta o processamento destes minérios através da rota pirometalúrgica convencional (WATLING, 2013), já que esta apresenta pouca flexibilidade para tratar concentrados com alto nível de impurezas deletérias, como flúor, cloro e arsênio.
Atualmente (2016), existe um desequilíbrio entre a oferta e a demanda de cobre no mundo e as estratégias para aumentar a oferta envolvem o processamento de minérios complexos, a reciclagem do metal a partir de sucatas eletrônicas, por exemplo, e o desenvolvimento de processos para extrair cobre de concentrados impuros (WATLING, 2013). Neste contexto, extensivas pesquisas no campo da hidrometalurgia têm sido conduzidas com o objetivo de desenvolver um processo eficiente para extrair cobre a partir de minérios ou concentrados calcopiríticos, porém sem grande sucesso (DEVI et al., 2000; WATLING, 2006; NAZARI et al., 2011). Isto ocorre porque, a
calcopirita é o mais estável dos sulfetos de cobre, sendo refratária à maioria dos tratamentos hidrometalúrgicos (CARNEIRO e LEÃO, 2007). Por esta razão, a dissolução de calcopirita é relativamente lenta e a extração do metal é baixa se comparada à de outros sulfetos de cobre como calcocita (Cu2S), bornita (Cu5FeS4) e covelita (CuS).
19 (H2SO4-NaCl) utilizando íons férricos (Fe2(SO4)3) ou íons cúpricos (CuSO4.5H2O) como agentes
oxidantes, com o intuito de investigar os aspectos relacionados à cinética de lixiviação de calcopirita. Posteriormente, foram realizados ensaios de transformação da calcopirita em outros sulfetos de cobre através da reação com enxofre gasoso, utilizando temperaturas no intervalo de 300 °C a 450 °C, seguida da lixiviação atmosférica dessas fases no mesmo meio citado anteriormente. Em virtude da caracterização dos produtos da reação entre calcopirita e enxofre gasoso, que mostrou que as fases formadas quando a temperatura estava acima de 400 oC eram
bornita (45%) e pirita (31%), estudos de lixiviação química de uma amostra de concentrado contendo como principal sulfeto de cobre o mineral bornita, foram incluídos ao trabalho para fins de comparação.
Em termos de organização este trabalho foi divido em cinco capítulos. O capítulo 2 apresenta uma revisão crítica dos aspectos relacionados à cinética de lixiviação dos sulfetos em questão, calcopirita e bornita, bem como os fundamentos da transformação da calcopirita em outros sulfetos de cobre.
O capítulo 3, aborda a cinética de lixiviação da calcopirita em meio sulfato-cloreto antes do processo de transformação e está apresentado no formato de um artigo. Este artigo foi publicado no periódico científico International Journal of Mineral Processing. Apesar de numerosos estudos
já terem sido realizados com o objetivo de determinar parâmetros cinéticos da lixiviação de calcopirita (YÉVENES et al., 2010), ainda existem divergências na literatura acerca deste assunto.
Por exemplo, em relação à etapa controladora, alguns autores (HIRATO et al., 1987; CÓRDOBA et al., 2008; KAPLUN et al., 2011; RUIZ et al., 2011) sugeriram controle por reação química para
a dissolução de calcopirita enquanto outros estudos observaram controle por difusão (MUNOZ et al., 1979; BONAN et al., 1981; CARNEIRO e LEÃO, 2007).
20 tamanho (LEVENSPIEL, 1999). Entretanto, em alguns casos, conclusões inconsistentes podem ser encontradas após aplicação do SCM quando a PSD da amostra é negligenciada (GBOR e JIA, 2004). No artigo publicado no International Journal of Mineral Processing, a incorporação da
PSD ao SCM foi necessária para que valores de energia de ativação aparente consistentes com controle da reação fossem determinados. Os experimentos foram conduzidos em meio sulfato-cloreto e a oxidação da calcopirita por íons cúpricos ou íons férricos foi comparada. A escolha do meio lixiviante foi feita levando-se em consideração que a escassez da água e o aumento do rigor legislação em relação ao seu uso, têm impulsionado a busca por processos metalúrgicos que utilizem água do mar ou salobra (WATLING, 2013).
No capítulo 4 são abordados os resultados obtidos pela reação entre a calcopirita e enxofre gasoso, denominada sulfidização, para produzir sulfetos de cobre lixiviáveis. Estes resultados também estão apresentados no formato de um artigo que foi publicado no periódico científico Metallurgical and Materials Transactions B. Este estudo partiu do princípio de que a transformação da
calcopirita em um mineral menos refratário à lixiviação pode ser uma alternativa para extração de cobre a partir de concentrados impuros, pois embora a sulfidização envolva uma etapa pirometalúrgica (temperaturas da ordem de 300 °C – 450 °C), a mesma é realizada em temperaturas bastante inferiores à da rota pirometalúrgica convencional (1200 °C), fato que tende a minimizar os problemas relacionados à emissão de poluentes.
Nesse sentido, alguns aspectos relacionados às fases formadas e quantificação das mesmas ainda não foram totalmente esclarecidos, apesar de algumas publicações das décadas de 1970-80 (PARKER et al., 1975; DEMOPOULOS e DISTIN, 1983) e mais recentemente Padilla et al.
21 Investigações sobre a lixiviação da bornita em meio ácido, mesmo a partir de concentrados/minérios contendo este mineral como fase principal são relativamente poucas e existem incertezas relacionadas ao mecanismo da reação, aos produtos gerados, bem como aos fatores que influenciam na dissolução desta fase (ACRES et al., 2010; MAJUSTE et al., 2012).
Levando-se em consideração que a bornita, além de ser um sulfeto de cobre de grande importância econômica, foi encontrada como produto da sulfidização, um artigo sobre a lixiviação de um concentrado de bornita foi incluído na tese. Este artigo foi submetido para publicação no periódico científico Chemical Engineering Journal.
Normalmente, nas investigações sobre a lixiviação da bornita reportadas na literatura são utilizados íons férricos (DUTRIZAC et al., 1970; PESIC e OLSON, 1983; MOSWEU, 2014) ou
oxigênio (PESIC e OLSON, 1984) como oxidantes. Recentemente, processos de lixiviação de sulfetos de cobre em meio clorídrico utilizando íons cúpricos como oxidantes, como HidroCopper e Intec, foram testados em escala industrial (LU e DREISINGER, 2013). Porém, estudos envolvendo especificamente a lixiviação da bornita utilizando íons cúpricos como oxidante não foram encontrados em domínio público. Por esta razão, no capítulo 5 foi abordada de maneira sistemática a lixiviação da bornita utilizando íons cúpricos como oxidantes em um meio sulfato-cloreto, tal como no artigo sobre a cinética de lixiviação da calcopirita abordado no capítulo 3. Porém, o modelo utilizado para descrever a cinética de lixiviação da bornita foi o modelo do grão. De acordo com este modelo, as partículas são constituídas de diversos grãos que possuem o mesmo formato e tamanho (SZEKELY et al., 1976; SOUZA et al., 2007b). O reagente fluido difunde
através dos interstícios dos grãos com o progresso da reação e os grãos reagem individualmente (GEORGIOU e PAPANGELAKIS, 1998).
Finalmente o capítulo 6 traz as considerações finais desta tese, abordando as principais conclusões, as contribuições originais ao conhecimento e as sugestões para trabalhos futuros.
1.2 Objetivos
22 tanto foi imprescindível avaliar a cinética de lixiviação do sulfeto original e do produto da reação, a bornita. Desta maneira, os objetivos específicos da presente tese foram:
(i) Estudar a cinética de lixiviação da calcopirita aplicando as equações propostas por Gbor e Gia (2004) em meio sulfato-cloreto;
(ii) Investigar a transformação da calcopirita em outro sulfeto a partir da reação com enxofre gasoso e definir parâmetros que influenciam na mesma;
(iii) Avaliar a cinética de lixiviação da bornita por íons Cu2+ em meio sulfato-cloreto;
(iv) Propor um mecanismo para lixiviação da bornita por íons Cu2+;
23
2 CAPÍTULO 2 - REFERENCIAL TEÓRICO
2.1 Parâmetros que influenciam na lixiviação de calcopirita
A oxidação da calcopirita por íons Fe3+, em um meio contendo apenas sulfato, é representada pela
reação 2.1 (DUTRIZAC, 1981; PALMER et al., 1981). Já em um meio híbrido, contendo tanto
íons sulfato quanto íons cloreto, além da reação 2.1, a oxidação da calcopirita por íons cúpricos também ocorre (equação 2.2) (BONAN et al., 1981; RUIZ et al., 2011).
CuFeS2 (s) + 4Fe3+ (aq) = Cu2+ (aq) + 5Fe2+ (aq)+ 2S0 (s) (2.1)
CuFeS2 (s) + 3Cu2+ (aq) = 4Cu+ (aq) + Fe2+ (aq) + 2S0 (s) (2.2)
É fato conhecido que a adição dos íons cloreto aumenta a velocidade de dissolução de sulfetos e também a extração de cobre em meio sulfato. Isso é atribuído principalmente a dois fatores: (i) a redução da passivação da calcopirita, devido ao aumento da porosidade da camada de produtos (MAJIMA et al., 1985; LU et al., 2000; CARNEIRO e LEÃO, 2007); (ii) complexação dos íons
cuprosos, adicionando um novo par redox ao sistema (CARNEIRO e LEÃO, 2007). A solubilidade dos íons cuprosos é baixa em sistemas aquosos sem a presença de um agente complexante. Porém, os íons cloreto são capazes de formar complexos estáveis com os íons cuprosos mantendo-os em solução, favorecendo assim a extração de cobre (BONAN et al., 1981; CARNEIRO e LEÃO,
2007). Portanto, em processos de lixiviação em meio clorídrico ou híbrido (sulfato-cloreto), os íons cúpricos podem ser o agente oxidante (BONAN et al., 1981; HIRATO et al., 1987;
WATLING, 2014).
Como o potencial de oxidação estabelecido pelo par Fe2+/Fe3+ não é particularmente alto, além da
24 por íons férricos pode ser atribuído à maior rapidez e reversibilidade do par Cu2+/Cu+ em relação
ao Fe3+/Fe2+ na superfície da calcopirita (DUTRIZAC, 1992).
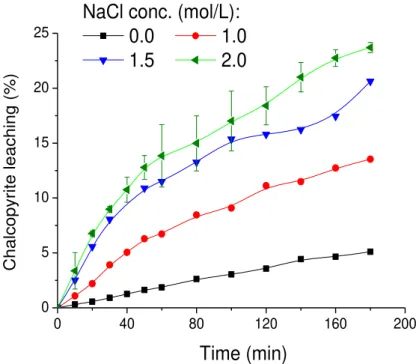
Não existe um consenso sobre até qual valor de concentração o íon cloreto influencia a extração de cobre a partir do referido sulfeto. Lu et al. (2000) e mais recentemente Ruiz et al. (2011), em
experimentos realizados em meio H2SO4-NaCl-O2,concluíram que a adição de cloreto de sódio
aumenta significativamente a velocidade de dissolução da calcopirita até concentrações de 0,5 mol/L de NaCl. Já Palmer et al. (1981), em experimentos realizados em meio clorídrico
utilizando íons férricos como oxidante, obtiveram acréscimos consideráveis na extração de cobre para adições de até 1,0 mol/L de NaCl. Carneiro & Leão (2007), utilizando solução lixiviante contendo ácido sulfúrico, íons férricos e oxigênio, observaram que a extração de cobre aumentou pela elevação da concentração de cloreto em concentrações de até 1,5 mol/L do ânion. Finalmente, Yévenes et al. (2010) ao avaliar o efeito da adição de íons cloreto, no intervalo de concentração
de aproximadamente 0,2mol/L a 2,0mol/L, em um meio contendo 0,2mol/L de HCl, 7,8mmol/L de Cu2+ e potencial químico mantido constante em 580mV pela aspersão de O
2/N2, observaram
que a concentração do ânion não influenciou no percentual de cobre dissolvido a partir da calcopirita.
Em relação à concentração inicial de oxidante no sistema, diferentes pesquisadores (MUNOZ et al., 1979; DUTRIZAC, 1981) têm apontado que a taxa de dissolução de calcopirita é fortemente
afetada pela concentração inicial de íon férrico, mas somente em concentrações do íon menores que 0,01 mol/L. Em concentrações elevadas, o efeito da variação da concentração do oxidante sobre a dissolução da calcopirita é insignificante (CÓRDOBA et al., 2008). Já quando os íons
cúpricos são utilizados como oxidante, existem contradições na literatura acerca da influência do aumento da concentração inicial destes íons no sistema na dissolução da calcopirita. Segundo Hirato et al. (1987), a dissolução da calcopirita varia linearmente com o tempo e é de meia ordem
em relação a concentração de íons cúpricos, no intervalo de 0,01 mol/L a 1,00 mol/L em temperatura de 67 °C. Estes resultados foram baseados em experimentos de lixiviação utilizando discos rotativos e meio lixiviante contendo 0,2 mol/L de HCl sob atmosfera de nitrogênio. Já Yévenes et al. (2010), ao realizar experimentos com partículas suspensas, meio lixiviante contendo
0,2 mol/L de HCl, potencial químico mantido constante em 580 mV em atmosfera de O2/N2 e
25 pequenas quantidades de íons cúpricos (1,6 mmol/L) é importante para se obter velocidades de lixiviação aceitáveis.
Outro parâmetro importante em processos hidrometalúrgicos é a temperatura, pois está diretamente relacionado com a cinética das reações envolvidas durante a etapa de lixiviação. Existe uma concordância entre os diversos autores de que o aumento na temperatura provoca um aumento substancial na velocidade de dissolução da calcopirita (DUTRIZAC, 1981; YÉVENES et al.,
2010; WATLING, 2014).
O aumento da velocidade de dissolução da calcopirita em função da temperatura foi observado em trabalhos utilizando diferentes meios lixiviantes, bem como oxidantes. Por exemplo, Yévenes et al. (2010) observaram um aumento na extração de cobre de 10% para 80% em 800 h de
experimento, quando a temperatura foi aumentada de 25 °C para 50 °C, em um meio lixiviante contendo 0,2 mol/L de HCl, 7,8 mmol/L de Cu2+ e potencial químico mantido constante em
580 mV pela aspersão de O2/N2. Lu et al. (2000), utilizando um meio híbrido sulfato-cloreto
(0,8 mol/L de H2SO4, 1 mol/L de HCl) e oxigênio como oxidante, mostraram que a extração de
cobre a partir da calcopirita aumentou de aproximadamente 30% para 95%, após 9 h de experimento, quando a temperatura foi aumentada de 60 °C para 95 °C. Por sua vez, Bonan et al.
(1981), utilizando íons cúpricos como oxidante, em um meio puramente clorídrico (0,1 mol/L de HCl e 3 mol/L de NaCl) e desareada, constataram um aumento de dez vezes, 5% para 50%, no percentual de cobre extraído ao aumentar a temperatura de 75 °C para 95 °C, em um período de três horas.
Como pode ser observado, a variação do percentual de cobre extraído em função da temperatura varia em função do meio e oxidantes empregados. Além disso, o intervalo de temperatura empregado pelos autores é diferente. Estes fatos dificultam a comparação dos dados obtidos em trabalhos distintos, porém em todos pode se verificar um efeito significativo do percentual de cobre extraído em função da elevação da temperatura.
26
2.2 Parâmetros que influenciam na lixiviação da bornita
O primeiro estudo sobre a lixiviação da bornita foi reportado Sullivan (1931), que realizou experimentos em garrafas mantidas em uma sala com temperatura controlada. Diversos parâmetros que influenciam a lixiviação foram avaliados pelo autor. Em relação ao tamanho partícula, apesar de analisar uma extensa faixa tamanhos, de 74 µm a 6730 µm, em solução contento 1% íons férricos (p/v, como sulfato férrico) e 0,5% de ácido sulfúrico, Sullivan (1931) observou que o tamanho da partícula não influenciou na extração de cobre a partir da bornita, ou seja, tanto para as partículas com grandes áreas superficiais, quanto para partículas maiores, as velocidades de reação foram muito parecidas. Vale ressaltar este comportamento foi observado para duas temperaturas diferentes, 35 ºC e 50 ºC.
O efeito do tamanho das partículas sobre a lixiviação da bornita também foi avaliado por Pesic e Olson (1983) em meio clorídrico (0,36 mol/L de HCl). Em experimentos realizados a 30 °C e com concentração de íon férricos 0,1 mol/L, quatro faixas granulométricas, de 74 µm +45 µm até -150 µm +104 µm, foram avaliadas. Os autores, assim como Sullivan (1931), não observaram influência da área superficial das partículas na velocidade de dissolução da bornita. Apenas, quando a concentração de íon férrico presente no meio lixiviante era de 0,005 mol/L, foi notada uma pequena dependência inversa do tamanho da partícula com a velocidade de dissolução. Logo, relatou-se que quando não existia quantidade suficiente de íon férrico, o tamanho da partícula interferiu na dissolução de cobre. Em experimentos realizados com temperaturas acima de 30 °C, não foi verificada influência do tamanho de partícula na dissolução da bornita em nenhuma das concentrações de íons férricos citadas.
27 Em relação à concentração de íons férricos, Sullivan (1931) observou que partículas com tamanho entre -150 µm +74 µm foram lixiviadas na mesma velocidade quando soluções contendo 0,5% de ácido de sulfúrico e 0,5%, 1% ou 2% de íons férricos foram empregadas. Em todos os casos aproximadamente 100% do cobre foi dissolvido após 12 dias em experimentos realizados a 35 °C. Dutrizac et al. (1970) também estudaram a lixiviação de bornita em solução ácida de sulfato
férrico, porém os experimentos foram conduzidos com bornita sintética sinterizada em discos rotativos. A solução utilizada por Dutrizac et al. (1970)foi mantida a 70 °C e continha 0,1 mol/L
de ácido sulfúrico com concentrações de íon férricos variando de 0,001 mol/L a 1 mol/L. Os autores observaram que a velocidade de dissolução da bornita foi dependente da concentração de íons férricos para concentrações menores do que 0,06 mol/L; a partir deste valor, a velocidade de dissolução de bornita não foi alterada pelo aumento da concentração de íons férricos. Segundo Pesic e Olson (1983), a velocidade de dissolução da bornita também não se mostrou dependente da concentração de íons férricos em meio clorídrico (0,36 mol/L HCl), quando os experimentos foram realizados com partículas de tamanho entre -150 µm +104 µm e temperatura superior a 30 °C. Portanto, existe um consenso entre autores de que concentração de íons férricos não tem efeito na lixiviação da bornita desde que existam íons férricos o suficiente para saturar a superfície do sólido (PESIC e OLSON, 1983). Em outras palavras, em concentrações elevadas, o efeito do aumento da concentração do oxidante sobre a dissolução da bornita é insignificante.
Assim como observado para a calcopirita, diversos autores relataram que a elevação da temperatura aumentou a velocidade de dissolução da bornita. Sullivan (1931) realizou experimentos de lixiviação de bornita com partículas de tamanho entre -150 µm +74 µm em soluções contendo 0,5% de ácido de sulfúrico e 1% de íons férricos em três diferentes temperaturas, 23 °C, 35 °C e 50 °C, e observou extrações de cobre de 47%, 68% e 98%, respectivamente, após 140 horas (5 dias) de experimento. Já Pesic & Olson (1983) avaliaram o efeito da temperatura em meio clorídrico, 0,36 mol/L de HCl – 0,08 mol/L de FeCl3, com
28
2.3 Mecanismos de lixiviação da bornita
A bornita é um sulfeto contendo 63,3% de cobre, 11,1% de ferro e 25,6% de enxofre, sua fórmula química pode ser representada por Cu5FeS4. Este sulfeto é frequentemente encontrado em minérios
de cobre juntamente com a calcocita (Cu2S) e calcopirita (CuFeS2), ocupando o terceiro lugar em
importância econômica (SULLIVAN, 1931; DUTRIZAC et al., 1970).
Dentre os reagentes mais importantes para a lixiviação dos sulfetos de cobre em pressão ambiente, destacam-se o ácido sulfúrico e sulfato férrico como sendo a combinação mais comumente empregada (WATLING, 2013). Porém, mesmo neste meio, estudos de dissolução da bornita são escassos na literatura e existem muitas incertezas sobre o mecanismo de oxidação desse mineral (ACRES et al., 2010; MAJUSTE et al., 2012).
Sullivan (1931), com o intuito determinar o mecanismo de lixiviação da bornita, realizou experimentos de lixiviação em garrafas mantidas em uma sala com temperatura controlada a 35 °C. Foi realizada a dissolução de partículas desse mineral com tamanho entre -150µm +74 µm, em uma solução ácida de sulfato férrico. O autor, então, propôs que a formula da bornita fosse escrita como 2Cu2S.CuS.FeS e sugeriu que a dissolução na presença de sulfato férrico ocorreria
de acordo com as equações 2.3, 2.4 e 2.5:
Cu2S + Fe2(SO4)3 = CuSO4 + 2FeSO4 + CuS (2.3)
CuS + Fe2(SO4)3 = CuSO4 + 2FeSO4 + S (2.4)
FeS + Fe2(SO4)3 = 3FeSO4 + S (2.5)
Estudos posteriores, como os realizados por Dutrizac et al. (1970), mostraram que o mecanismo
29 (Cu5-xFeS4) denominada “bornita não-estequiométrica”, sugerindo a sequência de equações 2.6 a
2.8 para descrever a dissolução da bornita:
Cu5FeS4 + xFe2(SO4)3 = Cu5-xFeS4 + xCuSO4 + 2xFeSO4 (2.6)
Cu5-xFeS4 + (4-x) Fe2(SO4)3 = CuFeS2 + (4-x) CuSO4 + (8-2x) FeSO4 + 2S (2.7)
CuFeS2 + 2Fe2(SO4)3 = CuSO4 + 5FeSO4 + 2S (2.8)
Segundo os autores (DUTRIZAC et al., 1970), as reações (2.6) e (2.7) são rápidas e a (2.8) é lenta,
caracterizando estágios distintos da lixiviação. Em experimentos realizados em temperaturas inferiores a 40 °C, os autores observaram que a única reação que ocorreu, foi a mostrada na equação 2.6, ou seja, nenhuma fase adicional, além da “bornita não-estequiométrica”, foi formada e a dissolução de cobre cessou após toda a transformação da bornita em “bornita não -estequiométrica” mesmo após decorridos 15 dias de experimento.
Price e Chilton (1981), ao estudar as reações anódicas de dissolução de bornita sintética em meio sulfúrico através de experimentos eletroquímicos em diferentes temperaturas, também verificaram a presença de dois estágios distintos de dissolução. Porém, esses estágios aconteceram tanto em alta (65 °C a 95 °C) quanto em baixas temperaturas (20 °C a 50 °C). Através de análises por microssonda eletrônica, os autores associaram a “bornita não-estequiométrica” à fase Cu2,5FeS4 e
ressaltaram que a natureza deste produto pode variar em função da presença de um oxidante. As equações 2.9 e 2.10, válidas para intervalo de temperatura de 20 °C a 70 °C, mostram a sequência de reações propostas pelos autores.
Cu5FeS4 = Cu2,5FeS4 + 2,5 Cu2+ + 5e- (2.9)
Cu2,5FeS4 = 2,5 Cu2+ + Fe3+ + 4S0 + 8e- (2.10)
30 63,35% de cobre e 11,4% de ferro, com tamanho de partícula entre -150 µm +104 µm, em solução ácida (0,36 mol/L de HCl) com cloreto férrico (0,1 mol/L de FeCl3), mantida em um reator a
temperatura constante (intervalo avaliado entre 0,5° C e 90° C), verificaram a presença de dois diferentes estágios de lixiviação, com um período de transição entre eles. O primeiro estágio foi reportado como extremamente rápido, terminando quando 28% do cobre foram extraídos. O período de transição ocorreu entre 28% e 40% de dissolução de cobre e foi caracterizado por ser consideravelmente lento. O segundo estágio, ainda mais lento, aconteceu a partir de 40% de extração de cobre com formação de enxofre elementar.
Através de difração de raios-X, os autores encontraram como produto, dos dois estágios, a calcopirita, mas análisesquímicas por microssonda eletrônica comprovaram que a fase tinha a fórmula química Cu3FeS4. Assim, a sequência de reação de dissolução da bornita proposta por
Pesic e Olson (1983) é dada pelas equações 2.11 e 2.12:
Cu5FeS4 + 4Fe3+ = Cu3FeS4 + 2Cu2+ + 4Fe2+ (Estágio I) (2.11)
Cu3FeS4 + 8Fe3+ = 3Cu2+ + 9Fe2++ 4S (Estágio II) (2.12)
Em outro trabalho, Pesic e Olson (1984) estudaram também a lixiviação da bornita, porém em meio sulfúrico utilizando como oxidante o oxigênio. Por causa da reação lenta com O2, foi possível
investigar apenas o primeiro estágio de dissolução e os resultados encontrados se diferenciaram em parte dos resultados obtidos quando foi utilizado o íon férrico como oxidante. Nos experimentos com oxigênio como oxidante, os referidos autores também encontraram o composto intermediário Cu3FeS4,porém junto com a covelita (CuS), que não foi encontrada como produto
quando a bornita foi lixiviada em meio férrico. Além disso, eles identificaram que primeiro aconteceu uma dissolução de ferro a partir da bornita, para depois, como tentativa de recuperar a estabilidade da estrutura, os íons cuprosos se dissolverem na solução, formando covelita na superfície da bornita. Utilizando a fórmula estrutural da bornita (Cu3FeS4)2-.2Cu+, os autores
propuseram as equações 2.13 e 2.14, para explicar a dissolução do ferro e formação da covelita, respectivamente:
31 x[(Cu3Fe▫S4)2- . 2Cu+] + x[(Cu3FeS4)2-. 2Cu+] = 4xCuS + xCu3FeS4 + 3xCu+ + 3xe- (2.14)
Onde, a fórmula química [(Cu3Fe▫S4)2- . 2Cu+] foi denominada bornita deficiente em ferro, na qual
o ponto representa a vacância deixada pelo íon férrico.
A sugestão de formação de covelita como produto da lixiviação da bornita foi reportada também por outros autores, como Dutrizac et al. (1985) e Bevilaqua et al. (2010). Dutrizac et al. (1985)
observaram a formação de covelita em um estudo detalhado sobre as fases formadas durante a lixiviação da bornita tanto em meio clorídrico (0,3 mol/L de HCl) quanto sulfúrico (0,3mol/L de H2SO4) contendo íons férricos (0,2 mol/L). Nos experimentos de lixiviação realizados a 10 °C,
utilizando tanto o FeCl3 quanto o Fe2(SO4)3, foi observada uma rápida dissolução de cobre que
cessou quando essa dissolução atingiu 26,5% de extração (em meio sulfato) ou 30% (em meio cloreto), resultando na “bornita não-estequiométrica” (Cu5-xFeS4). A caracterização dos resíduos
mostrou também que inicialmente (até 30 minutos de lixiviação) calcopirita e covelita eram formadas, e para tanto foram propostas as reações 2.15 e 2.16 para descrever o mecanismo de dissolução:
Cu5-xFeS4 = CuFeS2 + 2Cu(4-X)/2S (2.15)
2Cu(4-X)/2S+ (2-X)Fe3+ = (2-X)/2 Cu2+ + CuS + (2-X)Fe2+ (2.16)
A presença das fases calcopirita e covelita foi mais claramente observada nos experimentos utilizando amostras massivas de bornita (disco rotativo) do que em experimentos realizados com partículas suspensas (-150 µm +104 µm). Na superfície das partículas suspensas, com o progresso da reação, apenas a fase Cu3FeS4 foi detectada no resíduo. Este fato foi atribuído a uma dissolução
relativamente rápida das fases intermediárias calcopirita e covelita ou ocorrência de uma reação no estado sólido, conforme mostrado na equação 2.17 (DUTRIZAC et al., 1985):
32 Nesse mesmo estudo, em experimentos realizados em altas temperaturas (65 °C e 95 °C) foi observado que a fase Cu3FeS4 começava a se dissolver formando enxofre elementar. A extensão
desta dissolução, que ocorreu conforme representado pela equação 2.18, mostrou-se extremamente dependente da temperatura empregada.
Cu3FeS4 + 8Fe3+ = 3Cu2+ + 9Fe2+ + 4S0 (2.18)
Já Bevilaqua et al. (2010) detectaram a formação de covelita como produto intermediário em
estudos de biolixiviação de bornita por Acidithiobacillus ferooxidans. Esta fase foi observada tanto
nos experimentos não inoculados quanto nos inoculados, porém foi dissolvida apenas na presença dos micro-organismos. Sendo neste último caso observada também a formação de enxofre elementar como produto da reação. A formação de nenhum outro produto intermediário da dissolução da bornita foi mencionada pelos autores.
2.4 Cinética de lixiviação da calcopirita e bornita
A modelagem cinética de sistemas fluido-sólido é normalmente empregada para interpretar os resultados experimentais e para obter informações sobre os mecanismos de reação (GBOR e JIA, 2004). Dentro os modelos matemáticos, o modelo do núcleo não reagido (SCM) é bastante utilizado para descrever os sistemas sólido/fluido. Muitas aplicações do mesmo são encontradas nos campos da metalurgia extrativa, e os exemplos incluem a oxidação de sulfetos metálicos para produzir óxidos (ustulação), a redução de óxidos metálicos por gases redutores e a extração de metais a partir de minérios/concentrado por lixiviação. No estabelecimento deste modelo, considera-se que o reagente sólido é não poroso e está inicialmente cercado por um filme fluido através do qual ocorre a transferência de massa entre a partícula sólida e o fluido. Com o prosseguimento da reação, pode-se formar uma camada de cinza/inerte que fica aderida em torno do núcleo da partícula que ainda não reagiu e, assim, o tamanho da partícula é mantido constante (LEVENSPIEL, 1999). A reação prossegue no sentido “de fora para dentro” da partícula e pode ser representada de maneira geral pela equação 2.19:
33 No caso da presente tese, o sólido B representa a calcopirita ou a bornita e o reagente A o oxidante, íons Fe3+ ou Cu2+. Sendo a reação irreversível, os estágios que contribuem diretamente para
cinética da reação são os que se seguem: (i) difusão do reagente A através da camada limite para a superfície do sólido; (ii) difusão de A através da camada de cinza até a superfície do núcleo não reagido e (iii) reação de A com o sólido B. As resistências dos diferentes estágios costumam apresentar grandes variações. Em tais casos, pode-se considerar que o estágio com resistência mais alta seja o controlador da velocidade (LEVENSPIEL, 1999). Neste modelo assume-se ainda que as partículas são esféricas, reagem isotermicamente e a concentração do oxidante no líquido é constante ou em excesso (GBOR e JIA, 2004).
Sempre que a resistência devida à difusão através da camada limite tiver a maior magnitude, a relação entre a fração de B convertida em produtos (XB) e o tempo (t) é dada pela equação 2.20:
𝑋𝐵= 𝑘𝑚 𝑡 (2.20)
Onde:
𝑘𝑚 = 3𝑏𝑘𝑔𝐶𝐴𝑏/𝜌𝑟 (2.21)
b = mols de B consumido por mols de A reagido;
kg = coeficiente de transferência de massa de A na camada limite (m/s);
CAb = concentração de A no seio do líquido (mol/m3); ρ = densidade molar de B (mol/m3);
r = raio da partícula (m).
O controle por difusão através da camada limite pode ser desprezado quando a velocidade de agitação for selecionada de forma a eliminar a resistência associada a esta etapa do processo.
Para a situação na qual a resistência devida à difusão através da camada de cinza controla a velocidade da reação, a relação entre a fração de B convertida em produtos (XB) e o tempo (t) é
34 1 − 3(1 − 𝑋𝐵)
2
3+ 2(1 − 𝑋𝐵) = 𝑘𝑑 𝑡 (2.22)
Onde:
𝑘𝑑 =6𝑏𝐷𝜌𝑟𝑒2𝐶𝐴𝑏 (2.23)
b = mols de B consumido por mols de A reagido;
De = coeficiente de difusão efetiva de A através da camada de cinzas (m2/s);
CAb = concentração de A no seio do líquido (mol/m3); ρ = densidade molar de B (mol/m3);
r = raio da partícula (m).
Quando a reação química é o estágio controlador, a equação que relaciona a fração de B convertida em produtos (XB) e o tempo (t) é mostrada em 2.24:
1 − (1 − 𝑋𝐵) 1
3 = 𝑘𝑟 𝑡 (2.24)
Onde,
𝑘𝑟 = 𝑏𝑘1𝐶𝐴𝑏 𝑛
𝜌𝑟 (2.25)
b = mols de B consumido por mols de A reagido;
k1 = constante de velocidade da reação química (m(3n-2)/mol(n-1).s);
n = ordem de reação em relação a A;
CAb = concentração de A no seio do líquido (mol/m3); ρ = densidade molar de B (mol/m3);
r = raio da partícula (m).
Uma vez obtidos os valores de kd ou kr, a equação de Arrhenius pode ser aplicada para estimar a
dependência da velocidade de reação com a temperatura, que é expressa pela energia de ativação da reação (KAPLUN et al., 2011). A constante de velocidade está relacionada à temperatura
35 𝑘 = 𝑘0𝑒𝑥𝑝 (−𝑅𝑇) → 𝐿𝑜𝑔 𝑘 = 𝐿𝑜𝑔 𝑘𝐸𝑎 0−2,303 ∙ 𝑅𝑇 𝐸𝑎 (2.26)
k = constante de velocidade;
k0 = fator pré-exponencial da equação de Arrhenius;
Ea = energia de ativação;
R = constante real dos gases; T = temperatura.
O SCM foi aplicado no estudo da cinética de lixiviação de calcopirita tanto em meio sulfato e quanto meio cloreto para estimar o efeito da temperatura na velocidade da reação, além de auxiliar na conclusão de qual é a etapa controladora (KAPLUN et al., 2011). Para sistemas
hidrometalúrgicos, geralmente, assume-se que para controle químico tem-se Ea > 40 kJ/mol (>
10 kcal/mol), enquanto que para controle por difusão, tem-se Ea < 20 kJ/mol (< 5 kcal/mol).
Quando o valor da energia de ativação se encontra entre 20 kJ/mol e 40 kJ/mol há indícios que o controle da reação é misto.
No tocante à etapa controladora da lixiviação da calcopirita os dados reportados na literatura são bastante controversos, pois alguns autores concluíram que o processo é controlado pelo transporte de massa enquanto outros tem sugerido controle químico. Alguns estudos reportando controle por difusão são questionáveis, dado que a energia de ativação encontrada não é consistente com este tipo de controle (GBOR e JIA, 2004; YÉVENES et al., 2010).
Uma das causas que podem afetar a interpretação dos dados obtidos em experimentos de lixiviação é o fato destes sistemas possuírem múltiplas partículas que apresentam uma ampla distribuição de propriedades, como tamanho, composição mineralógica, entre outras, as quais influenciam no comportamento do sistema como um todo. Portanto, a cinética de um sistema constituído de múltiplas partículas, em alguns casos, não pode ser analisada da mesma maneira que seria analisada a cinética de um sistema de uma partícula individual (HERBST, 1979).
36 ao plotar a expressão 1 - (1-XB)1/3 versus tempo, o resultado obtido foi uma reta. Porém, este
comportamento só foi observado quando as partículas apresentavam uma faixa estreita de distribuição tamanho, já quando as partículas apresentavam uma ampla distribuição de tamanho, a “lei 1 - (1-XB)1/3” não era obedecida, o que era esperado dado que o SCM foi estabelecido
incialmente para partículas de mesmo tamanho inicial. Em sistemas com partículas com ampla distribuição de tamanhos, as partículas menores reagem mais rapidamente do que as maiores, afetando a velocidade de dissolução como um todo.
Dois parâmetros importantes utilizados para caracterizar a distribuição de tamanho de partículas são o tamanho médio, ou tendência central da distribuição, µ, e o coeficiente de variação (CV), ou amplitude de variação dos tamanhos em torno da média. Herbst (1979) ao comparar as curvas de fração reagida da calcopirita em função do tempo para diferentes valores de CV, observou, para elevados valores de CV, que a velocidade decrescia com o progresso da lixiviação. Este fato ocorre porque o aumento no valor de CV resulta em um aumento na abundância de partículas tanto de pequeno como de grande diâmetro (relativos a média) na amostra. As partículas pequenas reagem mais rapidamente, dando origem à forma inicial da curva, já as partículas maiores reagem mais lentamente sendo responsáveis pelo formato da curva ao final do processo. Embora Herbst (1979) tenha constatado que a exclusão da PSD no SCM poderia causar sérios erros na interpretação dos resultados, nenhuma uma análise quantitativa foi conduzida no sentido de incluir a mesma no SCM. Uma das formas de incorporação da PSD no SCM para cada um dos três regimes de controle cinético foi proposta por Gbor e Gia (2004) e será abordada no capítulo 3.
A maioria dos estudos reportados na literatura sobre cinética lixiviação de calcopirita negligenciam a PSD das amostras e esta pode ser a causa de obtenção de conclusões inconsistentes após aplicação do SCM. Por exemplo, desconsiderar a distribuição de tamanho de partículas pode resultar em mudança do ajuste da equação que representa o controle químico para controle por difusão na camada de cinza, quando o CV está entre 0,7 e 1,2 considerando-se o tamanho médio de partícula de 100µm e krn=0,1µm/min (GBOR e JIA, 2004).
37 Os correspondentes valores da média (µ) e coeficiente de variação de distribuição (CV) também são mostrados (HERBST, 1979).
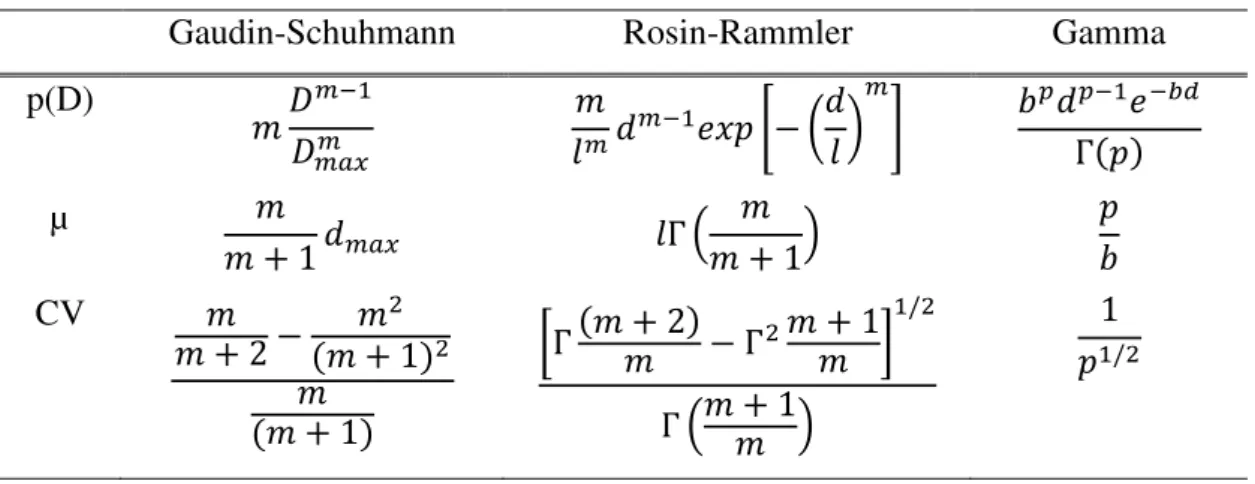
Tabela II. 1: Distribuição de tamanho de partículas comumente empregada (HERBST, 1979)
Gaudin-Schuhmann Rosin-Rammler Gamma
p(D)
𝑚𝐷𝐷𝑚−1 𝑚𝑎𝑥𝑚
𝑚
𝑙𝑚𝑑𝑚−1𝑒𝑥𝑝 [− ( 𝑑
𝑙 ) 𝑚
] 𝑏𝑝𝑑𝑝−1𝑒−𝑏𝑑
Γ(𝑝)
µ 𝑚
𝑚 + 1 𝑑𝑚𝑎𝑥 𝑙Γ (
𝑚 𝑚 + 1)
𝑝 𝑏
CV 𝑚
𝑚 + 2 − 𝑚
2 (𝑚 + 1)2 𝑚
(𝑚 + 1)
[Γ (𝑚 + 2)𝑚 − Γ2𝑚 + 1 𝑚 ]
1/2
Γ (𝑚 + 1𝑚 )
1 𝑝1/2
Em relação à energia de ativação para a dissolução da calcopirita, uma grande dispersão dos valores é reportada na literatura independente do meio ou oxidante utilizado. Por exemplo, encontram-se valores variando entre 38 kJ/mol e 130 kJ/mol, obtidos por diferentes autores, utilizando íons férricos como oxidante em meio sulfato, para temperaturas variando de 35 °C a 100 °C (DUTRIZAC, 1981; CÓRDOBA et al., 2008). Em meio clorídrico, utilizando íons férricos
como oxidante, são encontrados valores de energia de ativação na faixa de 1,1 kJ/mol a 87 kJ/mol, considerando o intervalo de temperatura de 3,5 °C a 110 °C (AL-HARAHSHEH et al., 2008). Já
em experimentos utilizando íons cúpricos como oxidante, também em meio clorídrico, os valores de energia de ativação encontrados variam de 35 kJ/mol a 134,7 kJ/mol, para temperaturas variando de 60 °C a 105 °C (BONAN et al., 1981; HIRATO et al., 1987; LUNDSTRÖM et al.,
2005).
Apesar do SCM ter sido aplicado em diversos trabalhos, encontram-se na literatura estudos que utilizaram outras formas para determinar as constantes de velocidade das reações de dissolução da calcopirita e, por conseguinte, a energia de ativação. Por exemplo, Dutrizac (1981) ao comparar a cinética de lixiviação de calcopirita, utilizando íons férricos como oxidante, em meio sulfato (0,2 mol/L FeSO4, 0,3 mol/L H2SO4) e clorídrico (0,2 mol/L FeCl3, 0,3 mol/L HCl) constatou que
38 75 kJ/mol e 63 kJ/mol, para os meios sulfato e clorídrico, respectivamente. Porém, segundo o próprio autor este método não possui um embasamento teórico forte.
Majima et al. (1985) ao realizar experimentos utilizando seções polidas de calcopirita expostas em
uma solução lixiviante contendo 0,2 mol/L de HCl e 0,1 mol/L de FeCl3 constaram que a
dissolução de cobre variou linearmente com tempo no intervalo de temperatura de 55 °C a 90 °C. A partir das inclinações das retas os autores determinaram as constantes de velocidade e obtiveram um valor de energia de ativação aparente de 69 kJ/mol. Levando-se em consideração o valor da energia de ativação foi sugerido que a dissolução da calcopirita, nas condições experimentais empregadas, apresentava um controle químico. Este resultado está de acordo com o resultado obtido por Al-Harahsheh et al. (2008), que também encontraram um valor de energia de ativação
aparente de 69 kJ/mol para o intervalo de temperatura de 70 °C a 90 °C, utilizando uma solução lixiviante contendo 0,5 mol/L de HCl e 0,5 mol/L de FeCl3. Porém, os últimos empregaram o SCM
para descrever a cinética do processo e obtiveram um bom ajuste dos dados experimentais para a equação de controle por reação química, com coeficientes de correlação (R2) superiores a 0,98.
Hirato et al. (1987) realizaram experimentos com discos rotativos, em um meio puramente
clorídrico (0,2 mol/L de HCl) e com íons cúpricos como oxidante (0,1 mol/L de CuCl2). Utilizando
o método das velocidades iniciais encontraram um valor de energia de ativação aparente de 81,5 kJ/mol para intervalo de temperatura de 60 °C a 85 °C e concluíram, suportados por experimentos eletroquímicos, que o controle da reação era químico. Já Bonan et al. (1981), ao
realizar experimentos com partículas de calcopirita suspensas em um meio puramente clorídrico (0,1 mol/L de HCl e 3 mol/L de NaCl), utilizando íons cúpricos como oxidante, observaram um bom ajuste dos dados experimentais ao SCM para controle por difusão na camada de cinzas. E, a partir do mesmo, determinaram uma energia de ativação aparente de 71 kJ/mol para o intervalo de temperatura de 85 °C a 104 °C. Portanto, sugeriram um controle de reação por difusão no intervalo de temperatura considerado. Conforme já discutido anteriormente, o valor de energia de ativação encontrado pelos autores é relativamente alto para este tipo de controle.
Yévenes et al. (2010) utilizando a faixa de dados em que extração de cobre variou linearmente
39 ativação para dois concentrados distintos (72 kJ/mol e 73 kJ/mol), e por conseguinte, concluíram que etapa limitante da velocidade da reação era uma reação química/eletroquímica na superfície do mineral.
Lu et al. (2000) utilizando a região (5 h iniciais de experimento) na qual a extração de cobre, a
partir de um concentrado de calcopirita, variou linearmente com o tempo para encontrar a constante de velocidade da reação, em um meio contendo 1 mol/L de NaCl e 0,8 mol/L de H2SO4,
utilizando oxigênio como oxidante, encontraram uma energia de ativação aparente de 48 kJ/mol em baixas temperaturas (60 °C e 70 °C) e 20 kJ/mol em elevadas temperaturas (85 °C e 95 °C), sugerindo que, em baixas temperaturas, o controle é principalmente químico e em temperaturas mais elevadas ocorre uma mudança no controle da reação para difusional. Entretanto, esta drástica mudança no valor da energia de ativação em um faixa relativamente estreita de temperatura parece pouco realista (HIRATO et al., 1987). Estes resultados são diferentes dos obtidos mais
recentemente por Ruiz et al. (2011) em meio lixiviante semelhante (1 mol/L de NaCl e 0,2 mol/L
de H2SO4) e utilizando oxigênio como oxidante. De acordo com estes últimos autores, os dados
experimentais obtidos apresentaram uma boa correlação com o SCM com controle químico para todas as temperaturas avaliadas, sendo determinada uma energia de ativação de 91,2 kJ/mol para o intervalo de temperatura de 80 °C a 100 °C, sugerindo um controle eletroquímico para a reação.
Segundo Dutrizac (1981), as diferenças observadas nos valores de energia de ativação são justificadas pelo fato de que a energia de ativação pode sofrer variações em função da faixa de temperatura estudada, do tamanho de partícula, da presença de impurezas e outros minerais de na amostra, da origem da amostra, concentração de reagentes e também do método aplicado para cálculo de velocidade. Além disso, o ajuste dos dados a determinada equação/modelo depende da região da curva utilizada para extrair os dados, do número de pontos utilizados e do tempo total da reação. Apesar das discrepâncias dos valores encontrados na literatura, deve-se salientar que as energias de ativação são mais elevadas em meio sulfato do que em meio de cloreto quando íons férricos são empregados como oxidante (CÓRDOBA et al., 2008).
Embora menos frequentes do que os estudos sobre a lixiviação da calcopirita, alguns trabalhos sobre a lixiviação da bornita reportaram a energia de ativação aparente desta reação (DUTRIZAC
40 ocorre em mais de um estágio. Com relação ao primeiro estágio de dissolução, em meio sulfato e utilizando bornita sintética sinterizada em discos, Dutrizac et al. (1970) observaram que a cinética
tinha tendência parabólica e, no intervalo de temperatura de 5 °C a 35 °C, encontraram um valor de 23 kJ/mol para energia de ativação. Como este valor é relativamente baixo, os autores sugeriram que a velocidade da reação era controlada pela difusão da solução de sulfato férrico pelos poros e trincas da “bornita não-estequiométrica” formada com o decorrer da reação. Em meio clorídrico, considerando o intervalo de temperatura de 0,5 °C a 30 °C, Pesic e Olson (1983) também encontraram uma energia de ativação na ordem de 25 kJ/mol, porém sugeriram um controle cinético misto para o primeiro estágio da lixiviação, considerando tanto a difusão dos íons férricos na camada limite quanto a reação química para formar o composto intermediário importantes na determinação da velocidade da lixiviação.
Para o segundo estágio de lixiviação, em meio sulfato, Dutrizac et al. (1970) encontraram para