w w w . r b h h . o r g
Revista
Brasileira
de
Hematologia
e
Hemoterapia
Brazilian
Journal
of
Hematology
and
Hemotherapy
Review
article
Hemoglobin
D-Punjab:
origin,
distribution
and
laboratory
diagnosis
Lidiane
de
Souza
Torres
∗,
Jéssika
Viviani
Okumura,
Danilo
Grünig
Humberto
da
Silva,
Claudia
Regina
Bonini-Domingos
UniversidadeEstadualPaulista(UNESP),SãoJosédoRioPreto,SP,Brazil
a
r
t
i
c
l
e
i
n
f
o
Articlehistory:
Received11September2014 Accepted27October2014 Availableonline23February2015
Keywords:
Hemoglobins Haplotypes Diagnosis
Hemoglobinopathies
a
b
s
t
r
a
c
t
ThisreviewdiscusseshemoglobinD-Punjab,alsoknownashemoglobinD-LosAngeles,one ofthemostcommonhemoglobinvariantsworldwide.Itisderivedfromapointmutation inthebeta-globingene(HBB:c.364G>C;rs33946267)prevalentinthePunjabregion, North-westernIndian.HemoglobinD-Punjabcanbeinheritedinheterozygosiswithhemoglobin Acausingnoclinicalorhematologicalalterations,orinhomozygosis,therarestformof inheritance,aconditionthatiscommonlynotrelatedtoclinicalsymptomatology.Moreover, thisvariantcanexistinassociationwithotherhemoglobinopathies,suchasthalassemias; themostnoticeableclinicalalterationsoccurwhenhemoglobinD-Punjabisassociatedto hemoglobinS.Theclinicalmanifestationsofthisassociationcanbesimilarto homozygo-sisforhemoglobinS.AlthoughhemoglobinD-Punjabisacommonvariantgloballywith clinicalimportanceespeciallyincasesofdoubleheterozygosis,hemoglobinS/D-Punjabis stillunderstudied.InBrazil,forexample,hemoglobinD-Punjabisthethirdmostcommon hemoglobinvariant.Thus,thispapersummarizesinformationabouttheorigin,geographic distribution,characterizationandoccurrenceofhemoglobinD-Punjabhaplotypestotry toimproveourknowledgeofthisvariant.Moreover,alistofthemaintechniquesused initsidentificationisprovidedemphasizingtheimportanceofcomplementarymolecular analysisforaccuratediagnosis.
©2015Associac¸ãoBrasileiradeHematologia,HemoterapiaeTerapiaCelular.Published byElsevierEditoraLtda.Allrightsreserved.
Introduction
Mutationsingenesencodinghemoglobinchainsarepresentin about7%ofworldwidepopulation.1Thesegeneticalterations
∗ Correspondingauthorat:InstitutodeBiociências,LetraseCiênciasExatas(IBILCE),UniversidadeEstadualPaulista(UNESP),RuaCristóvão Colombo,2265,JardimNazareth,15054-000SãoJosédoRioPreto,SP,Brazil.
E-mailaddress:[email protected](L.S.Torres).
can affect the production rate of globin chains and cause thalassemia,ortheycanmodifymoleculestructureand gen-eratehemoglobinvariants.1,2
Hemoglobinvariantsusuallyaretheconsequenceof sin-gle amino acid substitutions caused by point mutations
http://dx.doi.org/10.1016/j.bjhh.2015.02.007
in genes encoding globin chains, resulting in a tetramer withdifferentphysicochemicalcharacteristics.3Accordingto
theGlobinGeneServerdatabase(http://globin.cse.psu.edu/), 1198 hemoglobin variants were described until Septem-ber 2014. Most of the hemoglobin variants described do notcause symptomaticclinicalmanifestations;however, in somecases,theycanbeassociatedtorelevant pathophysi-ology,e.g.,hemoglobinS(Hb S).Hb Sisthe mostfrequent hemoglobinvariantintheworld;itsclinicaloutcomeissevere inhomozygousorinassociationwithotherrelatively com-monhemoglobinopathies,suchasbeta-thalassemia,HbCor HbD.3,4 Differenttotheotherhemoglobinopathies,thislast
oneisstillpoorlystudied,especiallyinBrazil,wherethereare fewrecentstudiesaboutitsprevalence,diagnosisand clini-calaspects.Moreover,HbDpresentsconsiderablegeographic distributionandcanbeassociatedwithHbS,forminga het-erozygouscompositewithpeculiarclinicalseverity.
Thus,thisstudyaimedtogatherinformationavailablein the literature about type D hemoglobins, morespecifically aboutHbD-Punjab,inordertodrawattentiontoits impor-tance and to encourage the scientific community to take interestinthesubject,particularlyinBrazilwherestudieson thisvariantarescarce.
History
Untiltheearly1950s,onlyfourtypesofhemoglobinhadbeen describedincludingadulthemoglobin(HbA)andtwovariants (HbSandHbC).ThefourthidentifiedhemoglobinwasHbD whichwasfirstdescribedbyItanoin1951.Inhisstudy,Itano was analyzing a multiethnic family from the Los-Angeles regionwithBritish,AmericanandIndianfeatures,andfound amoleculewithcharacteristicsdifferenttotheotherknown hemoglobins.Thishemoglobinhadelectrophoreticmobility similartoHbSinalkalinepH,however,inacidpH,its migra-tion resembledHb A. Furthermore,Hb D exhibited normal solubility when in its reduced state and did not undergo sickling.5,6
Sincethediscoveryofthisnewvariantandtheknowledge ofitsmolecularstructure,otherhemoglobinswithsimilar pat-ternsdiscoveredinpopulationsfromdifferentethnicitieshave beennominatedaccordingtotheregionsinwhichtheywere described.Inastudyperformedin1962,Baglionianalyzedthe molecularstructureoffivehemoglobins,HbD-Chicago,Hb D-North Carolina,Hb D-Punjab,Hb D-Portugal and HbD-Oak Ridgeandfoundthatallexhibitedthesamechemical com-positionasthefirstonediscovered,HbD-LosAngeles.6,7The
most frequentdenominations forthis mutanthemoglobin foundintheliteratureareHbD-LosAngelesorHbD-Punjab.
Mutationˇ121Glu→Gln(GAA→CAA)
Hb D-Punjab is a variant derived from a point muta-tion in the beta-globin gene (HBB) in the first base of the 121 codon (GAA→CAA) with the substitution of glu-tamineforglutamicacid(Glu>Gln)inthebetaglobinchain.8
According tothe Globin Gene Server database, besides Hb D-Punjab, there are seven other types of Hb D, caused by different point mutations in HBB. They are Hb D-Agri (HBB:c.29C>A;364G>C), Hb D-Bushman (HBB:c.49G>C),
HincII
HincII
HinfI HinfI
AvaII
ψβ
ε
γ
Gγ
Aδ
β
BamHI
HincII
HindIII
HindIII
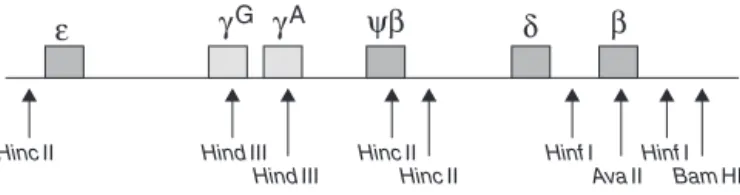
Figure1–Restrictionpolymorphicsitesincluster
beta-globinforhaplotypesscreeninginassociationwithHb D-Punjab
Source:ModifiedfromYavarianetal.19
Hb D-Ouled Rabah (HBB:c.60C>A or 60C>G), Hb D-Iran (HBB:c.67G>C), Hb D-Granada (HBB:c.68A>T), Hb D-Ibadan (HBB:c.263C>A)andHbD-Neath(HBB:c.365A>C).
Originandgeographicdistribution
HbD-Punjabisoneofthemostcommonhemoglobinvariants worldwide,afterHbSandHbC.ItisprevalentinPunjabregion, NorthwestIndian, withan estimatedfrequencyof2.0%.In westernIndia,morespecificallyintheGujaratregion,its fre-quencydropsbyonehalf.9
HbD-PunjabisalsocommonincountriessuchasItaly,10
Belgium,11Austria12andTurkey.13HbD-Punjabisthesecond
mostcommonhemoglobinalterationinTurkey;itoccursin 0.2%of thepopulation ofDenizli provincein southeastern Turkey,whereitisthemostcommonvariant.Atotalof57.9% ofabnormalhemoglobinsobservedinthisprovinceareHb D-Punjab,followedbyHbS,with21.9%.13 Asimilarfrequency
occursinXinjiangprovince,northwesternChina,where55.6% ofvarianthemoglobinsareHbD-Punjab.14,15
In Brazil there are no recent studies reporting the fre-quencyofHbD-Punjabinthepopulation,butinalargesurvey conductedin1993byBonini-Domingos,whichevaluatedthe prevalence of hemoglobinopathies in about 100,000 Brazil-ianindividualsfromeverystateinthecountry,HbD-Punjab was observed to be the third most common hemoglobin variant in the population.16 Posteriorly, in a screening for
hemoglobinopathies performedinahospital-based popula-tioninsoutheasternBrazilbySonatietal.,theauthorsfound hemoglobinalterationsin34.4%oftheindividuals,ofwhich 0.4%correspondedtoHbD-Punjab.Despitethelowfrequency, HbD-Punjabwasalsothethirdmostfrequentvariant identi-fiedinthispopulation.17
Initially,thegeographic distributionofHbD-Punjab sug-gests thatthismutationoriginated inthecentralregionof Asia, as it is prevalent in Punjab, a region of India, and NorthwesternChina.Thenitspreadtoneighboringcountries by migration.11,18,19 However, haplotype analysis of
beta-globinclustersprovidesanotherinsightintotheoriginofthis hemoglobin.Theanalysisofpolymorphicsites,5′-HincII,␥G HindIII,␥AHindIII,HincII,3′-HincII,5′-HinfI,AvaII,3′-
HinfI,3′-BamHI(Figure1),showedcertainhaplotypes associ-atedtoHbD-Punjab,suggestingamulticentricoriginforthis mutation.19–24
The study ofAtalay et al. considered sevenbeta-globin genepolymorphicsites(5′-HincII, ␥G HindIII,␥AHindIII,
Table1–HaplotypesdescribedintheliteratureassociatedwithHbD-Punjabandthelocationswheretheywerefirst found.
Polymorphicsitesofclusterbeta Occurrence
5′-
HincII
␥G
HindIII
␥A
HindIII

HincII
3′-
HincII
5′-
HinfI

AvaII
3′-
HinfI
3′-
BamHI
+ − − − − NR + NR + Mexico,18,20Italy,10,18
Iran18,21
+ − − − − NR + + NR Turkey,18,22India19
− + + − + NR + NR + Thailand8,18
− + + − + NR + + NR Turkey18
− + − − + NR + + NR Turkey18
− − + − + NR + + NR Turkey22
+ − − − − + NR + NR Iran,theNetherlands,
Belgium17
+ − − − − − NR + NR Iran17
− + − − + + NR + NR Iran17
− + − + + + NR + NR Iran,India,England17
NR:notreported.
haplotypes. Oneofthem isthe MediterraneanHaplotypeI (+−−−−++)whichisthemostcommonandfoundin popu-lations of Mexico,22 Italy10 and Iran.23 A second recently
discoveredpattern wasassociatedtoHbD-Punjab casesin Thailand(−++−+++)8andthethirdhaplotypewasfoundin Turkishpopulations(−+−−+++).20
In2009,Yavarianetal.analyzedsevenpolymorphicsites (5′-HincII,␥GHindIII,␥AHindIII,HincII,3′-HincII,5′-HinfI and3′-HinfI)andfoundfourhaplotypepatternsassociated toHbD-PunjabintheIndianpopulation.Thefirstandmost commonwas theMediterraneanHaplotype I(+−−−−++), thesecondseemstobeaproductoftheMediterranean Hap-lotypeIvariationprobablyduetoanindependentmutation (+−−−−−+), the thirdwas the same haplotype found in Turkey(−+−−+++),andthefourthacommonhaplotypein IndianandEnglishpopulations(−+−++++).19
Consideringthe same restrictionsites assessed by Ata-lay et al. in 2007and in 2009, Bahadiret al. reported the occurrenceofanother haplotypeinthe Turkishpopulation (−−+−+++).20,24 Due to lack of studies in Chinese and Indianpopulations,Pateletal.evaluatedindividualswithHb D-Punjab from Agharia inIndia and foundanother haplo-type(−−−−−++)inadditiontohaplotypeI(+−−−−++).21
Table1summarizesalltheseresults.
As the haplotypes described in association with Hb D-Punjabareprevalentindifferentregions,itissuggestedthat theoriginofthemutationincodon121maybe multicen-tric.AccordingtoYavarianetal.,oneimportantaspectisthat the mutationsiteis locatedina shortpalindromic region, wheremanymutationsaredescribed,suggestingthatcodon
121exhibitsahighermutationratethanisexpected.Thus, there is a chance that mutational events give rise to the same hemoglobin indifferent regions.19 However,it is not
yet possible to confirm the origin and distribution of this hemoglobinastherearefewstudiesabouthaplotypesin popu-lationsfromChina20andfromothercontinentssuchasNorth,
Southand Central America and Africa.In Brazil,no study hasbeen performedtoscreen haplotypesassociatedtoHb D-Punjab.
InheritanceofHbD-Punjabandassociatedclinical features
The Hb D-Punjab can be inherited in heterozygosis with normalHbA,characterizingtheheterozygoustrait.This con-ditionpresentsnoclinicalorhematologicalalterations.The homozygous component Hb DD, the rarestform of inheri-tance, isnot commonlyrelated tosymptomatic cases, but occasionallyindividualswiththisprofilecandevelopmildto moderatehemolyticanemia.25,26
The association of this variant with other abnormal hemoglobins,suchasHbSorthalassemiacanalsooccur. Usu-ally,interactionsbetweenHbD-Punjabandbetathalassemia coursewithmildmicrocyticandhypochromicanemia,butdo notpresentrelevantclinicalorhematologicalchanges.27
However, when Hb D-Punjab is associatedto Hb S, the doubleheterozygousHbS/D,theresultismoderatetosevere clinicalmanifestations;thisassociationcanbeclinically simi-lartohomozygousHbSS.25Pain,duetovaso-occlusiveevents,
is one of the most common complications. Furthermore, strokeandacutechestsyndromecanoccurincarriersofthis genotype.28–30
Recently,Pateletal.publishedastudyonHbS/D-Punjab patients with moderate to severe clinical symptomatology similar to the Hb SS genotype who had vaso-occlusion episodesorrequiredbloodtransfusions.Theauthorsobserved greatervulnerabilityforredcelllysisinHbS/D-Punjabpatients thanwiththeHbSSgenotype.31ClinicalseverityinHb
S/D-PunjabindividualscouldberelatedtothefactthatHbDfavors polymerizationofHbS.32
AS
A
B
C
D
+
–
+
–
–
+
Hb A
Hb S/D
Hb A2
Hb A/D
βA γ
A
γG βC
αA βS/βD
βC αA
βA/βD
Hb S Hb C
AD AS AC AD AS AC AC AD AC AD
+
–
Figure2–ElectrophoreticmobilityofHbD-Punjabcomparedtootherknownhemoglobins,suchasHbA,SandC.Images correspondtohemoglobinelectrophoresisincelluloseacetate(A),hemoglobinelectrophoresisinagarosegel(B),
polypeptidechainselectrophoresisincelluloseacetate(C)andpolypeptidechainselectrophoresisinpolyacrylamidegel(D). Thegenotypesofeachsampleareatthetopofthefigure.Currentdirectionandpositionofthedifferenthemoglobinsare besideeachmethod.
theaminoacidchangesinthe121Glu→Glnposition encour-agetheinteractionofHbSmoleculesandconsequentlyits polymerization.33,34Adachietal.showedthattheHbS
poly-merizationrateishigherwhenHbD-Punjabispresent.This mayexplaintheclinicalseverityofsicklecelldiseasein dou-bleheterozygousHbS/D-Punjab,asHbD-Punjabincreasesthe probabilityofsicklingduetoashortertransittimeofthese cellsinthebloodcapillaries.32
Hydroxyurea(HU)administrationisapotential therapeu-ticstrategyforsicklecellanemia(HbSS),showingefficacyin increasingHbFlevelsandconsequentlyreducingthenumber ofpainfulcrises,acutechestsyndrome,transfusions,and hos-pitalizations.However,HUtherapyforHbS/D-Punjabhasnot beenestablished.Pateletal.treatedHbS/D-Punjabpatients using low doses (10mg/kg/day) of HU noticed an effective reduction in the incidence of vaso-occlusion and the fre-quencyofbloodtransfusions.TheseresultssuggestthatHU isapromisingtherapyforclinicalsymptomatologyinHb S/D-Punjabindividuals.31
Laboratorydiagnosis
Numerous laboratory methods are used to diagnose hemoglobinvariantsandthalassemias.
Hemoglobinscanbeseparated byelectrophoresisin cel-luloseacetateatpH8.635andinagarosegelatpH6.2.36In
celluloseacetate,themigrationofHbDisslowerthanHbA withapositionsimilartoHbS,butinacidpHitsmigration positionisthesameasHbA.Theseelectrophoreticpatterns, however,arenotspecificforvariantsoftheHb Dgroup,as thereare other hemoglobinvariants withsimilar behavior, suchasHbGandHbKorle-bu.
Thestructuralcharacterizationofhemoglobinvariantsby electrophoreticseparationofhemoglobinpolypeptidechains isusedincasesinwhichtheresultsofthe initialtestsare inconclusive.37,38ThemutantDchainmigratessimilartothe
S chainusingthismethod,but slightlybelowthis fraction
inalkalinepH,whileitmigratestothesamepositionasthe normalAchaininacidpH(Figure2).
Itis alsopossibleto separatehemoglobins accordingto their isoelectric point (pI)by isoelectricfocusing (IEF).39 In
thecaseofHb D-Punjab,itspIisneartopH7.1,aposition similar toother hemoglobin variants such as Hb Korle-Bu (Figure 3). Althoughit hasgreat resolution, this technique requiresspecifickitsandismoreexpensivethanconventional electrophoresis.
Hb A
Hb F
Hb D-Ouled rabah
Hb D-Punjab
Hb Korle Bu
Hb S
Hb D-Iran
Meta-hemoglobin
Hb A2 pl 7.4
pl 7.3 pl 7.2 pl 7.1 pl 7.0
Hb C
Analyte ID
A
C
D
B
Time F Unknown P3 Ao Unknown Unknown A2 S-window Unknown F Unknown P2 P3 Unknown Ao A2 S-window 0.5 1.1 3.0 5.3 2.3 50.0 2.0 35.8 1.11 1.24 1.36 1.72 2.12 2.60 3.66 4.21 1 2 0.9 0.4 0.3 1.8 1.5 1.5 3.1 45.8 44.7 1.09 1.23 1.80 2.14 2.37 2.80 3.68 4.21 4.52 19102 9184 5273 36983 31704 31149 68206 941803 920453 9452 20009 56137 98985 43972 942058 40734 674234 1 2 3 4 % Total area 0.9% 30% 20% 10% 0 1 0 2 F A2 43 5 6
30% 20% 10% 0 1 0 5.218 4.232 1.957 2.325 2.667 4.782 Peak 1 2 3 4 5 6 7 8 9 10 11 12 13 0.420 0.538 0.653 0.783 0.990 1.192 1.957 2.325 2.667 3.810 4.232 4.782 5.218 0.415 2.662 2.973 3.730 4.247 4.788 5.202 5.630 0.28 0.69 0.77 0.97 1.10 0.85 0.93 1.00 0.8% 1.1% 1.1% 0.7% 1.2% 3.1% 47.5% 44.5% 5638 7389 7107 4872 8315 20676 318819 299037 671853 1 2 3 4 5 6 7 8 F A A A A S S S 0.30 0.38 0.47 0.56 0.71 0.85 1.39 1.66 0.70 1.00 1.11 0.85 0.93 0.5% 0.4% 0.8% 0.6% 0.2% 2.7% 0.2% 2.3% 3.0% 50.1% 0.7% 2.7% 35.7% 5015 4616 8685 6234 2442 27681 2452 24412 31117 521847 7326 27789 372058 1041674 F F F F F F F F A A A S S
RT Rel RT % Conc
Total area :
Total area :
Area
Peak RT Rel RT % Conc Area
5.202 5.630 0.415 2.662 2.973 3.730 4.247 4.788 Comment Comment A2 peak
Consistent with D-los angeles Consistent with S A1c peak
2 A0 peak
A2 peak
Consistent with D-los angeles 3.810 2
F A2
3 4 5 6
A2 F
F 3.1%
2063857 Total area
0.5% A2 2.0% 1885581 Area Analyte ID % Time Area
mm N HET N HET HET HET HET
564bp
296/268bp
Figure5–Agarosegelvisualizationoffragmentsgenerated bythePCR-RFLPtechniqueforscreeningof121Glu→Gln (GAA→CAA)mutation.Normalallelesgeneratetwo fragmentsandmutantallelesmaintainthe564bp fragment,indicatedbythearrows.N:absentmutation; HET:heterozygousforHbD-LosAngeles;mm:molecular markerof100bp.Restrictionenzyme:EcoRI(G↓AATTC).
High performance liquid chromatography (HPLC) meth-odshavebeendevelopedforhemoglobinfractionationwith high sensitivity and specificity. This technique is an inte-gratedmethodbasedonion-exchangetoseparateandidentify the relative percentage of specific hemoglobins in whole bloodsamples(Figure4).Anothermethodemployedtodetect hemoglobinfractionsiscapillaryelectrophoresis(CE)basedon theprincipleofelectrokineticseparation.Despitethe excel-lentresolutionandreproducibility,thistechniqueisonlynow arrivinginsomecountries,includingBrazil.
Reverse-phase HPLC (RP-HPLC) ofhuman globin chains has been reported as an important tool for detecting hemoglobinopathies. Nevertheless, although it is widely used for the detection, quantification and purification of many proteins from biological materials, its clinical appli-cation for hemoglobins is still limited due to the long analyticaltime,complexsamplepreparationandresolution problems.40,41 Recently,studieshavehadsuccessin
optimi-zingthemethod,40,41butinmanylaboratoriesthetechnique
isstillemployedexclusivelyforresearch.
AlthoughallthesetestsaimtodiagnoseHbD-Punjab,they arenotconsidered conclusivebecauseofthelargenumber ofhemoglobinvariantsthat are similartoeach other. Itis alwaysrecommendedtoperformmolecularanalysisfor geno-typeconfirmationwithPCR-RFLPasanalternative42(Figure5).
When the Hb D variant is not identified by the above-mentionedmethodologies,sequencingofbeta-globingeneor othertechniquesthatdiscriminate aminoacid changesare recommended.
Usingthesemethodologiesoverthelast10years,70cases of type D hemoglobins from southeast Brazil were diag-nosedintheHemoglobinandHematologicalGeneticDiseases LaboratoryoftheUniversidade Estadual Paulista(UNESP) a referralcenterforhemoglobindiagnosis.Ofthese,66(94.3%) hadaprofilecompatibletoHbD-Punjab,eight(12.1%)were compoundheterozygousHb S/D-Punjaband58 (87.9%) het-erozygous Hb A/D-Punjab, all ofwhich were confirmed by moleculartests. Theother four remaining casespresented electrophoreticandchromatographicprofilescompatiblewith the“HbA/D-non-LosAngeles”trait,possiblyHbA/D-Iran(data notpublished).
Conclusion
Althoughnotalwaysassociatedwithrelevantclinicalhistory, HbD-Punjabisarelativelycommonhemoglobinworldwide; it is the thirdmostcommon hemoglobin variantinBrazil. GreaterattentionisgivenwhentheassociationofHbD-Punjab withanotherabnormalhemoglobinorthalassemialeadsto clinicalsymptoms. However,this issueisstillunderstudied especiallyinBrazilandthepathophysiologicalmechanisms causedbyHbD-Punjabarepoorlyknown.Thereisnorecent informationabouttheprevalenceofHbD-Punjab,association withother hemoglobinvariants,or characterizationof typ-icalhaplotypes inthe Brazilianpopulation. Therefore, it is importanttoconductstudiesaboutthishemoglobinvariant.
Conflicts
of
interest
Theauthorsdeclarenoconflictsofinterest.
Acknowledgments
The authors would like to thank Fundac¸ão de Amparo à PesquisadoEstadodeSãoPaulo(FoundationforResearch Sup-portoftheStateofSaoPaulo)–FAPESP,bysupportingresearch inourlaboratory(processnumber2012/19653-1).Inaddition, tothankLucasPereiraRamosforEnglishtextrevision.
r
e
f
e
r
e
n
c
e
s
1.WeatherallDJ,CleggJB.Inheritedhaemoglobindisorders:an increasingglobalhealthproblem.BullWorldHealthOrgan. 2001;79(8):704–12.
2.KohneE.Hemoglobinopathies:clinicalmanifestations, diagnosis,andtreatment.DtschArzteblInt.
2011;108(31/32):532–40.
3.ThomCS,DicksonCF,GellDA,WeissMJ.Hemoglobin variants:biochemicalpropertiesandclinicalcorrelates.Cold SpringHarbPerspectMed.2013;3(3):a011858.
4.FrenettePS,AtwehGF.Sicklecelldisease:olddiscoveries,new concepts,andfuturepromise.JClinInvest.2007;117(4):850–8.
5.ItanoHA.Athirdabnormalhemoglobinassociatedwith hereditaryhemolyticanemia.ProcNatlAcadSciUSA. 1951;37(12):775–84.
6.VellaF,LehmannH.HaemoglobinDPunjab(DLosAngeles).J MedGenet.1974;11(4):341–8.
7.BaglioniC.Abnormalhumanhaemoglobins.VII.Chemical studiesonhaemoglobinD.BiochimBiophysActa. 1962;59(2):437–49.
8.FucharoenS,ChangtrakunY,SurapotS,FucharoenG, SanchaisuriyaK.MolecularcharacterizationofHbD-Punjab
121(GH4)Glu→GlninThailand.Hemoglobin.
2002;26(3):261–9.
9.KöselerA,ÖztürkO,AtalayA,AtalayEÖ.Allelefrequencyof VNTRlocusD1S80observedinHbD-LosAngelescarriers.Mol BiolRep.2012;39(12):10747–50.
10.FiorettiG,DeAngiolettiM,PaganoL,LacerraG,ViolaA,de BonisC,etal.DNApolymorphismsassociatedwithHbD-Los Angelesb121(GH4)Glu→GlninSouthernItaly.Hemoglobin.
1993;17(1):9–17.
121(GH4)Glu→GlnintheprovinceofLiegeBelgium.
Hemoglobin.1986;10(6):587–92.
12.LischkaA,PollakA,BauerK,AschauerH,BraunitzerG. HemoglobinD“LosAngeles”inanAustrianfamily: biochemicalidentification,clinicalaspects,andkindred study.Hemoglobin.1984;8(4):353–61.
13.AtalayEÖ,KoyuncuH,TurgutB,AtalayA,YildizS,BahadirA, etal.HighincidenceofHbD-LosAngeles121(GH4)Glu→Gln
inDenizliprovinceAegeanregionofTurkey.Hemoglobin. 2005;29(4):307–10.
14.LiHJ,ZhaoXN,QinF,LiHW,LiL,HeXJ,etal.Abnormal hemoglobinsinthesilkroadregionofChina.HumGenet. 1990;86(2):231–5.
15.PandeyS,MishraRM,PandeyS,ShahV,SaxenaR.Molecular characterizationofhemoglobinDPunjabtraitsand
clinical–hematologicalprofileofthepatients.SaoPauloMedJ. 2012;130(4):248–51.
16.Bonini-DomingosCR.Prevenc¸ãodashemoglobinopatiasno Brasil–DiversidadeGenéticaeMetodologiaLaboratorial. 144f.Tese(DoutoradoemCiências)–InstitutodeBiociências, LetraseCiênciaExatas.SãoJosédoRioPreto,SP:Univ. EstadualPaulista–Unesp;1993.
17.SonatiMF,KimuraEM,GrottoHZ,GervasioSA,CostaFF. Hereditaryhemoglobinopathiesinapopulationfrom southeastBrazil.Hemoglobin.1996;20(2):175–9.
18.BrittenhamGM.Globingenevariantsandpolymorphismin India.In:WinterWP,editor.Hemoglobinvariantsinhuman populations.Florida:CRCPressInc.;1988.p.79–110.
19.YavarianM,KarimiM,ParanF,NevenC,HarteveldCL, GiordanoPC.MultiCentricoriginofHbD-Punjab
121(GH4)Glu→Gln,GAA>CAA.Hemoglobin.
2009;33(6):399–405.
20.AtalayEÖ,AtalayA,UstelE,YildizS,OztürkO,KöselerA, etal.GeneticoriginofHbD-LosAngeles121(GH4)Glu→Gln,
GAA→CAAaccordingtothe-globingeneclusterhaplotypes. Hemoglobin.2007;31(3):387–91.
21.PatelDK,MashonRS,PatelS,DashPM,DasBS.-Globingene haplotypeslinkedwiththeHbD-Punjab121(GH4)Glu→Gln,
GAA→CAAmutationineasternIndia.Hemoglobin.
2010;34(6):530–7.
22.PereaFJ,Casas-Casta ˜nedaM,Villalobos-ArámbulaAR, BarajasH,AlvarezF,CamachoA,etal.HbD-LosAngeles associatedwithHbSor-thalassemiainfourMexican Mestizofamilies.Hemoglobin.1999;23(3):231–7.
23.RahimiZ,AkramipourR,NagelRL,AhmadiAS,MeratA, BahrehmandF.The-globingenehaplotypesassociatedwith HbD-LosAngeles121(GH4)Glu→GlninWesternIran.
Hemoglobin.2006;30(1):39–44.
24.BahadirA.HbD-LosAngelesbeta121(GH4)Glu→GnandHb
Beogradbeta121(GH4)Glu1Val:implicationsfortheir laboratorydiagnosisandgeneticorigins.TurkJHematol. 2009;26(1):17–20.
25.AdekileA,Muah-AlIA,AkarNA.DoeselevatedhemoglobinF modulatethephenotypeinHbSD-LosAngeles?Acta Haematol.2010;123(3):135–9.
26.TaghaviBasmanjM,KarimipoorM,AmirianA,JafarinejadM, KatouzianL,ValaeiA,etal.Co-inheritanceofhemoglobinD
and-thalassemiatraitsinthreefamilies:clinicalrelevance. ArchIranMed.2011;14(1):61–3.
27.NaoumPC,MoraesMS,RadispielJ,CavalheriPP,ValeriFF.Hb D/Talassemiabetaassociadaàanemiacrônica.RevBras HematolHemoter.2002;24(1):51–2.
28.Ohene-FrempongK,WeinerSJ,SleeperLA,MillerST,Embury S,MoohrJW,etal.Cerebrovascularaccidentsinsicklecell disease:ratesandriskfactors.Blood.1998;91(1):288–94.
29.CastroO,BrambillaDJ,ThoringtonB,ReindorfCA,ScottRB, GilletteP,etal.Theacutechestsyndromeinsicklecell disease:incidenceandriskfactors.Thecooperativestudyof sicklecelldisease.Blood.1994;84(2):643–9.
30.HamidiehAA,JaliliM,KhojastehO,GhavamzadehA.First reportofsuccessfulstemcelltransplantationinapatient withsicklecellhemoglobinDdisease.JPediatrHematol Oncol.2010;32(5):397–9.
31.PatelS,PurohitP,RanjeetSM,DehuryS,MeherS,SahooS, etal.Theeffectofhydroxyureaoncompoundheterozygotes forsicklecell-hemoglobinD-Punjab—asinglecentre experienceineasternIndia.PediatrBloodCancer. 2014;61(8):1341–6.
32.AdachiK,KimJ,BallasS,SurreyS,AsakuraT.Facilitationof HbSpolymerizationbythesubstitutionofGluforGlnat
121.JBiolChem.1988;263(12):5607–10.
33.CharacheS,ConleyCL.Rateofsicklecellsduring
deoxygenationofbloodfrompersonswithvarioussickling disorders.Blood.1964;24:25–48.
34.BeneschRE,KwongS,BeneschR,EdaljiR.Locationandbond typeofintermolecularcontactsinthepolymerizationof haemoglobinS.Nature.1977;269(5631):772–5.
35.Marengo-RoweAJ.Rapidelectrophoresisandquantitationof haemoglobinsoncelluloseacetate.JClinPathol.
1965;18(6):790–2.
36.VellaF.Acid-agargelelectrophoresisofhumanhemoglobins. AmJClinPathol.1968;49(3):440–2.
37.SchneiderRG.Differentiationofelectrophoretically hemoglobins–suchasS,D,GandPorA2,C,E,andO–by electrophoresisoftheglobinchains.ClinChem.
1974;20(9):1111–5.
38.AlterBP,GoffSC,EfremovGD,GravelyME,HuismanTH. Globinchainelectrophoresis:anewapproachtothe determinationoftheG/Aratioinfetalhaemoglobinandto studiesofglobinsynthesis.BrJHaematol.1980;44(4):527–32.
39.NaoumPC.Eletroforese–TécnicaseDiagnósticos.SãoPaulo: LivrariaSantosEditoraLtda;1990.
40.NematiH,BahramiG,RahimiZ.Rapidseparationofhuman globinchainsinnormalandthalassemiapatientsby RP-HPLC.MolBiolRep.2011;38(5):3213–8.
41.WanJH,TianPL,LuoWH,WuBY,XiongF,ZhouWJ,etal. Rapiddeterminationofhumanglobinchainsusing reversed-phasehigh-performanceliquidchromatography.J ChromatogrBAnalytTechnolBiomedLifeSci.
2012;15(901):53–8.
42.SaikiRK,GelfandDH,StoffelS,ScharfSJ,HiguchiR,HornGT, etal.Primer-directedenzymaticamplificationofDNAwitha thermostableDNApolymerase.Science.