Arq Neuropsiquiat r 2001;59(4): 932-935
FAM ILIAL CREUTZFELDT-JAKOB DISEASE ASSOCIATED
WITH A POINT M UTATION AT CODON 210 OF THE
PRION PROTEIN GENE
Nancy Huang
1, Suely K.N. Marie
2, Fernando Kok
2, Ricardo Nit rini
3ABSTRACT - Creut zfeldt -Jakob disease (CJD), t he most know n human prion disease, is usually sporadic but approximat ely 15% of t he cases are familial. To dat e, seven CJD cases w it h codon 210 mut at ion (GTT t o ATT) have been reported in the literature. We describe a case of a 57 year-old w oman w ho presented gait disturbances and rapidly progressive dement ia, leading t o deat h four mont hs aft er onset . Elect roencephalogram revealed periodic act ivit y, diffusion-w eight ed magnet ic resonance imaging show ed hypersignal in basal ganglia, and t est for 14-3-3 prot ein w as st rongly posit ive in t he CSF. The complet e prion prot ein gene coding region w as sequenced aft er PCR amplificat ion, show ing a point mut at ion in codon 210. This is t he first case of CJD w it h codon 210 mut at ion diagnosed in Brazil. We emphasize t he role of genet ic search for prion prot ein gene mut at ion, even in pat ient s present ing clinical feat ures resembling sporadic CJD.
KEY WORDS: familial Creut zfeldt -Jakob disease, prion prot ein gene mut at ion, codon 210, 14-3-3 prot ein.
Doença de Creutzfeldt-Jakob familial com mutação pontual no codon 210 do gene da proteína priônica
RESUM O - A doença de Creut zfeldt -Jakob (DCJ), a mais conhecida das doenças priônicas, é usualment e esporádica, mas cerca de 15% dos casos são familiais. Set e casos de DCJ familial com mut ação no codon 210 (GTT→ATT) foram relat ados na lit erat ura at é o present e moment o. Nós descrevemos o caso de uma mulher de 57 anos com dist úrbios de marcha e demência rapidament e progressiva, evoluindo para óbit o em 4 meses. Elet roencefalograma revelou at ividade periódica, ressonância magnét ica com t écnica de difusão most rou hipersinal em gânglios da base e t est e para prot eína 14-3-3 no líquido cefalorraqueano foi fort ement e posit ivo. A região codificadora do gene da prot eína priônica foi sequenciada após a amplificação por PCR, revelando mut ação de pont o no codon 210, const it uindo-se no primeiro caso com est a mut ação diagnost icado no Brasil. Enfat izamos a import ância da invest igação de mut ações do gene da prot eína priônica, mesmo em pacient es com quadro t ípico de DCJ esporádica.
PALAVRAS-CHAVE: doença de Creut zfeldt -Jakob familial, codon 210, gene da prot eína priônica, mut ação, prot eína 14-3-3.
Behavioral and Cognit ive Neurology Unit and Laborat ory for Neurologic Invest igat ions, Depart ment of Neurology, Facult y of M edicine, Universit y of São Paulo, São Paulo SP, Brazil: 1Post Graduat e St udent ; 2Assist ant Professor; 3Associat e Professor.
Received 4 June 2001, received in final form 17 August 2001. Accept ed 29 August 2001.
Dra. Nancy Huang - Rua Fernão Dias 279/14 - 05427-010 São Paulo SP - Brasil.
Creut zfeldt -Jakob disease (CJD) is t he most w ell
know n human prion disease, a group of diseases
relat ed t o t he presence of t he pat hologic prion
pro-t ein (PrP
Sc) in brain t issue
1. The incidence of CJD is
approximat ely 1 case per million inhabit ant s
annu-ally. The clinical pict ure includes a rapidly
progressi-ve dement ia, myoclonus, and pyramidal, ext
rapyra-midal or cerebellar syndromes
2. Elect
roencephalo-gram (EEG) may show periodic act ivit y in
approxi-mat ely 65% of cases, w it h a specificit y of 86%
3,4.
The presence of 14-3-3 prot ein in t he cerebrospinal
fluid (CSF), t oget her w it h suggest ive clinical feat ures,
has been considered highly sensit ive and specific for
diagnosis
4,5. How ever, definit ive diagnosis is st ill
de-pendent on pat hological confirmat ion of t he
pres-ence of PrP
Scin brain t issue.
About 15% of CJD cases are familial, associat ed
w it h point mut at ions or insert ions in t he prion
pro-t ein gene, w hich is locapro-t ed in pro-t he shorpro-t arm of
chro-mossome 20. The clinical feat ures and neuropat
ho-logical findings t end t o vary, depending on t he
dif-ferent mut at ions. Codon 129 polymorphysms are
also linked t o CJD risk, clinical p henot yp e and
prognosis
6-8.
Arq Neuropsiquiat r 2001;59(4) 933
CASE
A 57-year old w om an, born in Germ any and residing in Brazil, developed dizziness w hich w as soon f ollow ed by gait and speech dist urbances. A consult ant neurolo-gist det ect ed a global cerebellar syndrom e so request ed laborat ory t est s and a brain CT-scan t hat on exam inat ion proved unrem arkable. Her gait det eriorat ed and a rapidly progressive cognit ive decline associat ed w it h m yoclonic jerks becam e evident . A rout ine brain M RI w as report ed as norm al. When she w as f irst seen by us, t w o m ont hs aft er t he onset of her sympt oms, she w as alert but unable t o f ollow sim ple com m ands, w it h no eye-blinking t o t hreat ening st im uli, w hile react ing t o painf ul st im uli by sim ply w it hdraw ing t he st im ulat ed segm ent of body. She manifest ed spont aneous and st art le myoclonus, increased t one w it h cogw heel rigidit y in t he arm s, bilat eral grasp-ing ref lex, and Babinski sign in t he right f oot . Rout ine blood t est s and CSF analysis w ere w it hin norm al lim it s.
One of her sist ers had died one year earlier, in Ger-many, w it h neuropat hologically confirmed CJD. As far as w e have been informed, no ot her members of t he family have been affect ed by similar disease.
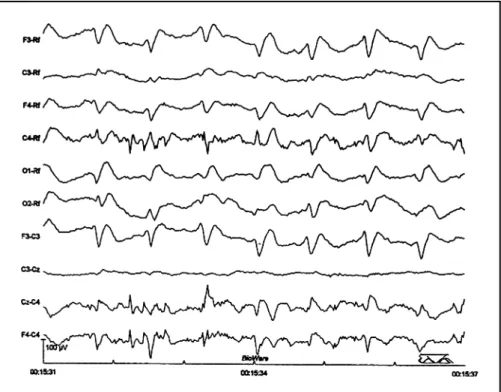
EEG revealed 1Hz periodic sharp w aves (Fig 1). M RI show ed mild high signal in t he st riat um on T2-w eight ed images, w hereas diffusion-w eight ed M RI (DWI) show ed high signal in t he st riat um, cort ical areas of t he insulas, left t emporal lobe, and t he borderland bet w een front al and pariet al lobes, bilat erally.
The 14-3-3 prot ein immunoassay in CSF w as performed as described by Zerr et al.10. 15 µl of CSF w ere applied t o
West ern Blot . Det ect ion of t he bound polyclonal ant ibody t o t he β-isoforms of the 14-3-3 protein (Santa Cruz Biotech, USA) w as performed using t he enhanced
chemilumines-cence det ect ion kit (Amersham, USA). A posit ive cont rol from t he NIH3T3 cell line and a negat ive cont rol w ere run on every gel. For t his pat ient , 14-3-3 prot ein w as posit ive in t he CSF sample (Fig 2).
Genom ic DNA w as ext ract ed f rom p erip heral b lood leukocyt es b y st and ard m et hod s11 f ollow ing t he
in-f orm ed consent oin-f in-f am ily m em bers. The com plet e PRNP cod ing reg ion w as seq uenced b y ob t aining t w o f rag
-Fig 1. EEG show ing periodic sharp w aves at 1 Hz.
Fig 2. Immunoassay for 14-3-3 prot ein in CSF. CSF from pat ient w it h V210I mut at ion (P) show s a band corresponding t o 14-3-3
934 Arq Neuropsiquiat r 2001;59(4)
Fig 3. PRNP coding region sequence: let t er “ N” indicat es a point mut at ion causing a subst it ut ion of valine by isoleucine in codon 210 (GTT-ATT).
m ent s af t er PCR am plif icat ion. The f ragm ent f rom open reading f ram e t o codon 134 w as am plif ied, using t he pair of primers FA (5'-CTGACGTTCTCCTCTTCATTTTG-3') and RA (5'-CTCATGGCACTTCCCAGCATGTA-3'). The cycle con-dit ions w ere set at s 94°C f or 30sec, 63°C f or 30sec , and 72°C f or 1 m in. The PCR react ions w ere carried out f or 30 cycles. The f ragm ent f rom codon 107 t o f inal of PRNP coding region w as am plif ied, using t he pair of prim ers FB (5 '- A A CCA A CATGA A GCA CATGG- 3 ') an d RB(5 '-TCCCTCAAGCTGGAAAAAGA-3'). The cycle cond it ions w ere set at 94°C f or 30 sec, 55°C f or 30 sec and 72 °C f or 1 m in. The PCR react ions w ere carried out f or 35 cycles. We f ound a point m ut at ion causing a subst it ut ion of va-line by isoleucine in codon 210 (GTT-ATT) (Fig.3) and het e-rozigosit y at codon 129 of t he prion prot ein gene.
The pat ient died four mont hs aft er onset of t he symp-t oms. Ausymp-t opsy w as nosymp-t performed.
DISCUSSION
Familial CJD associat ed w it h V210I mut at ion w as
first report ed by Ripoll et al.
12, and by Pochiari et al.,
in 1993
13. Including t hese original cases, a t ot al of 7
such cases w it h t his mut at ion have been report ed in
t he lit erat ure t o dat e: one French pat ient
12, one It
al-ian family w it h t w o cases
13, one Japanese pat ient
14,
t w o Chinese cases from t he same family
15and one
individual from Nort h Africa
16.
The clinical and neuropat hological feat ures
de-scribed in t he It alian, French and Japanese pat ient s
w ere very similar t o t hose of sporadic CJD w it h a
rapid evolut ion t o deat h (m ean, 4.1 m ont hs)
12-14.
How ever, t he It alian case had a new 24-bp delect ion
in t he ot her allele
13. One of t he Chinese pat ient s
pre-sent ed w it h panencephalit is at t he age of 48 years,
and t he disease durat ion exceeded 24 mont hs
15. The
pat ient from Nort h Africa present ed sensory
symp-t oms as symp-t he firssymp-t manifessymp-t asymp-t ion, a finding symp-t hasymp-t is
pro-minent in t he new variant of CJD(nvCJD)
17, and died
7.5 mont hs aft er t he onset of t he disease
16.
There have also been report s of individuals
bear-ing V210I mut at ion w ho had remained asympt
om-at ic om-at 72 and 82 years of age
13,16, show ing an
in-complet e penet rance.
Polym orphism at codon 129 is linked t o bot h
suscept ibilit y t o prion diseases, and t o dif f erent
clinical f eat ures
8. For exam p le, all nvCJD cases
report ed so far w ere homozygous for met hionine at
codon 129
.17, and homozygosit y at t his codon is a
predisposing risk factor to for sporadic and iatrogenic
CJD. In different mut at ions of t he prion prot ein gene,
129 polymorphism has been described as a fact or
t hat could modify survival t ime
18. The influence of
129 polymorphism on 210 mut at ion phenot ype has
not been est ablished
8. Five out of t he seven cases
w it h t he 210 m ut at ion described t o dat e, w ere
met hionine homozygous at t he codon 129
13-16. One
of t he It alian cases and t he French pat ient did not
undergo 129 polymorphism det erminat ion.
Arq Neuropsiquiat r 2001;59(4) 935
w it h t ype 1 PrP
Scand met hionine homozygosit y at
codon 129. This being charact erized by a rapidly
progressive dement ia w it h myoclonus and periodic
sharp w aves on EEG
19,20.
M RI in t his pat ient show ed t he t ypical findings
of high signal in basal ganglia and in a few cort ical
areas t hat have been described in CJD, and w hich
are more evident in diffusion-w eight ed M RI
9,21.
Recent ly, 14-3-3 prot ein in t he CSF w as described
as an import ant marker of CJD
4. In t his pat ient , t he
st rong posit ive react ion of t he 14-3-3 prot ein is
possibly relat ed t o t he rapid and progressive dest
ruc-t ion of ruc-t he brain ruc-t issue.
We emphasize t he import ance of genet ic invest
i-gat ion t o search for prion prot ein gene mut at ions,
even in pat ient s present ing t ypical clinical feat ures
resembling sporadic CJD.
REFERENCES
1. Prusiner SB. Prio ns (No bel lecture). Pro c Natl A cad Sci (USA ) 1998;95:13363-83.
2. Brandel JP, Delasnerie-Lauprêtre N, Laplanche JL, et al. Diagnosis of Creutzfeldt-Jakob disease: effect of clinical criteria on incidence esti-mates. Neurology 2000;54:1095-1099.
3. Steinhoff BJ, Räker S, Herrendorf G, et al. Accuracy and reliability of periodic sharp w ave complexes in Creutzfeldt-Jakob disease. A rch Neurol 1996;53:162-165.
4. Poser S, Mollenhauer B, Kraubeta A, et al. How to improve the clinical diagnosis of Creutzfeldt-Jakob disease. Brain 1999;122:2345-2351. 5. Kenney K, Brechtel C, Takahashi H, et al. An enzyme-linked
immu-nosorbent assay to quantify 14-3-3 proteins in the cerebrospinal fluid o f susp ected Creutzfeld t-Jako b d isease p atientes. A nn N euro l 2000;48:395-398.
6. Zerr I, Schulz-Schaeffer WJ, Poser S, et al. Current clinical diagnosis in Creutzfeldt-Jakob disease: identification of uncommon variants. Ann Neurol 2000;48:323-329.
7. Kovacs GG, Head MW, Bunn T, et al. Clinicopathological phenotype of codon 129 valine homozygote sporadic Creutzfeldt-Jakob disease. Neuropathol Appl Neurobiol 2000;26:463-472.
8. Mastrianni JA . The prion diseases: Creutzfeldt-Jakob, Gerstmann-Sträussler-Scheinker, and related disorders. J Geriatr Psychiatry Neurol 1998;11:78-97.
9. Nitrini R, Mendonça RA, Huang N, et al. Diffusion-weighted MRI in tw o cases of familial Creutzfeldt-Jakob Disease. J Neurol Sci 2001; 184:163-167.
10. Zerr I, Bodemer M, Gefeller O, et al. Detection of 14-3-3 protein in the cerebrospinal fluid supports the diagnosis ofd Creutzfeldt-Jakob dis-ease. Ann Neurol 1998;43:32-40.
11. Sambrook J, Fritsch EF, Maniatis T, et al. Molecular cloning: a labora-tory manual, vol 2. 2.Ed. Cold Spring Harbor NY: Cold Spring Harbor Laboratory Press, 1989.
12. Ripoll L, Laplanche JL, Salzmann M, et al. A new point mutation in the prion protein gene at codon 210 in Creutzfeldt-Jakob disease. Neurol-ogy 1993;43:1934-1938.
13. Pocchiari M, Salvatore M, Cutruzella F et al. A new point mutation of the prion protein gene in Creutzfeldt-Jakob disease. A nn Neurol 1993;34:802-807.
14. Furukaw a H, Kitamoto T, Hashiguchi H, et al. A Japanese case of Creutzfeldt-Jakob disease with a point mutation in the prion protein gene at codon 210. J Neurol Sci 1996;141:120-122.
15. Shyu W, Hsu Y, Kao M, et al. Panencephalitic Creutzfeldt-Jakob disea-se in a Chinedisea-se family: unusual predisea-sentation with PrP codon 210 muta-tion and identificamuta-tion by PCR-SSCP. J Neurol Sci 1996;143:176-180.
16. Mouillet-Richard S, Teil C, Laplanche JL et al. Mutation at codon 210 (V210I) of the prion protein gene in a North A frican patient w ith Creutzfeldt-Jakob disease. J Neurol Sci 1999;168:141-144.
17. Will RG, Z eidler M, Stew art GE, et al. Diagnosis of new variant Creutzfeldt-Jakob disease. Ann Neurol 2000;47:575-582.
18. Baker HF, Poulter M, Crow TJ, et al. Aminoacid polymorphism in hu-man prion protein and age at death in inherited prion disease. Lancet 1991;337:1286.
19. Parchi P, Castellani R, Capellari S, et al. Molecular basis of phenotypic variability in sporadic Creutfeldt-Jakob Disease. A nn Neurol 1996; 39:767-778.
20. Parchi P, Giese A, Capellari S, et al. Classification of sporadic Creutz-feldt-Jakob disease based on molecular and phenotypic analysis of 300 subjects. Ann Neurol 1999;46:224-233.