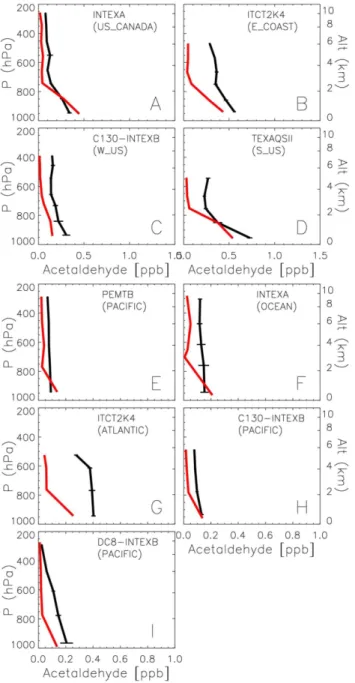
Global atmospheric budget of acetaldehyde: 3-D model analysis and constraints from in-situ and satellite observations
Texto
Imagem
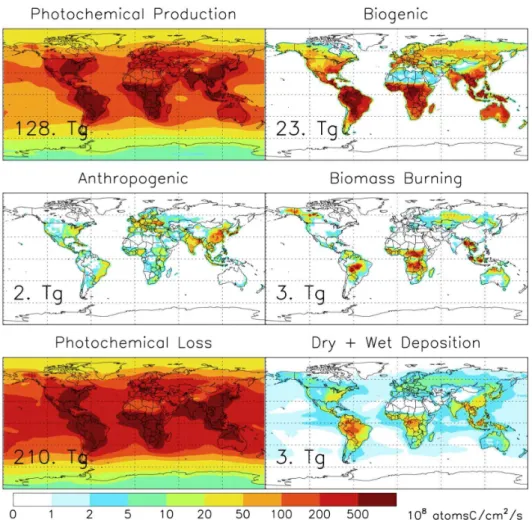
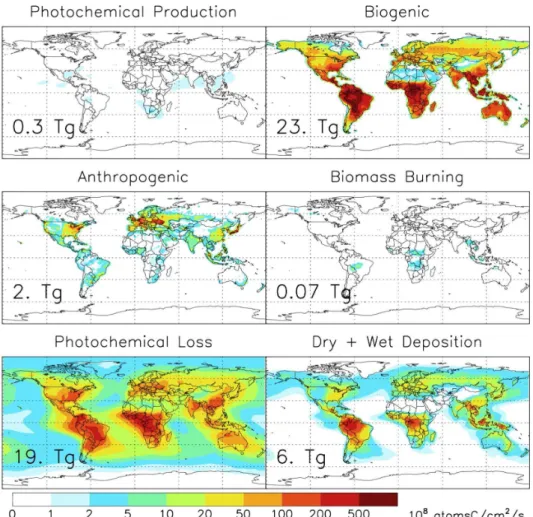
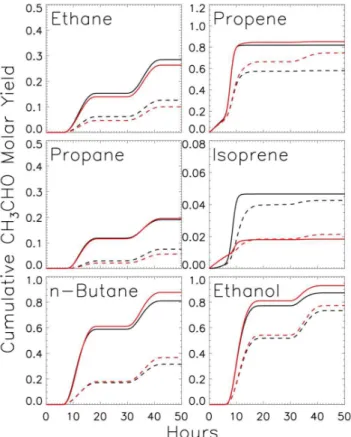

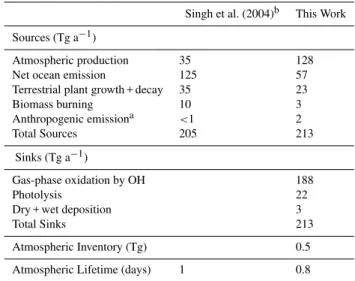

Documentos relacionados
Tidal analysis of 29-days time series of elevations and currents for each grid point generated corange and cophase lines as well as the correspondent axes of the current ellipses
The global levels of emissions in 2025 and 2030 were estimated by adding the estimated aggregate emission levels resulting from the implementation of the
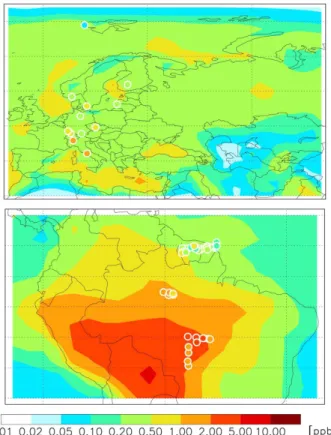
al., 2009, 2010) to constrain a 3-D global chemical trans- port model (CTM; GEOS-Chem) simulation of the volcanic aerosol distribution and direct radiative forcing following the
The structure of the remelting zone of the steel C90 steel be- fore conventional tempering consitute cells, dendritic cells, sur- rounded with the cementite, inside of
Here we present an explicit global simulation of BrC in a global 3-D chemical transport model (GEOS-Chem), including global BrC emission estimates from primary (3.9 ± 1.7 and 3.0 ±
However, alcohol sensitivity, amount of alcohol consumption and smoking status may not affect acetaldehyde production in mouth air, as participants refrained from smoking and from
To estimate the impact of aldehyde emissions at an airport on the air quality, formaldehyde and acetaldehyde concentrations were measured in the idle and taxiway areas of
This is likely due to the high amount of iridium and low amount of tin in this proportion because the iridium led the ethanol oxidation reaction to produce acetaldehyde, while
