Faculdade de Farmácia
A
VALIAÇÃO DA CITOTOXICIDADE E GENOTOXICIDADE
DA GLICIDAMIDA EM CÉLULAS MAMÁRIAS HUMANAS
Susana Isabel Casaca Figueiredo Bandarra
Mestrado em Controlo de Qualidade e Toxicologia dos Alimentos
Faculdade de Farmácia
A
VALIAÇÃO DA CITOTOXICIDADE E GENOTOXICIDADE
DA GLICIDAMIDA EM CÉLULAS MAMÁRIAS HUMANAS
Susana Isabel Casaca Figueiredo Bandarra
Mestrado em Controlo de Qualidade e Toxicologia dos Alimentos
Dissertação orientada por:
Professor Doutor Nuno Guerreiro de Oliveira
Professor Doutor Jorge Francisco Gaspar
ii
Inicio esta tese com o expressar do meu sincero agradecimento às pessoas e entidades que de algum modo contribuíram para a sua realização e sem as quais este trabalho não teria sido possível.
Em primeiro lugar, estou especialmente agradecida ao Prof. Doutor Nuno Guerreiro de Oliveira, meu orientador, pela sua vasta perspicácia e conhecimento transmitido. Pela sua hábil direcção no caminho correcto, permitindo-me ao mesmo tempo crescer e aprender com as suas críticas e sugestões pertinentes efectuadas durante a sua excelente orientação. Obrigada por todo o seu opoio.
Gostaria também de agradecer ao Prof. Doutor Jorge Francisco Gaspar, meu co-orientador, pelos seus sábios conselhos, pelos úteis comentários e recomendações sobre este trabalho e pela sua disponibilidade e partilha de conhecimento.
À equipa de Professores da Faculdade de Farmácia, Universidade de Lisboa, que leccionaram no ano curricular deste Mestrado a que me propus, pela fundamental contribuição com a transmissão de conhecimentos, em particular aqueles que me despertaram a curiosidade para este tema. Em especial, agradeço à Professora Matilde Castro pela sua excelente coordenação do Mestrado e notável capacidade de motivação, incentivo e encorajamento, proporcionando-me a mim e aos meus colegas um agradável desafio de aprendizagem.
Aos colegas do Mestrado pelo espírito de equipa que criámos e por tornarem este caminho muito mais fácil e agradável.
Ao Research Institute for Medicines and Pharmaceutical Sciences (iMed.UL) e, em especial, ao Chemical Biology and Toxicology Group (CBT) da Faculdade de Farmácia, onde desenvolvi a maior parte do meu trabalho, por me ter disponibilizado as instalações e os meios necessários à realização do mesmo.
Agradeço ainda o excelente ambiente de trabalho e ajuda que me proporcionaram todos os membros do laboratório, bem como a disponibilidade por parte de todas as pessoas com quem tive oportunidade de trabalhar, no CBT. Em especial, não posso deixar de agradecer às Doutoras Ana Sofia Fernandes e Joana Miranda pela imprescindível ajuda no trabalho laboratorial, pelo apoio incondicional, bem como pela disponibilidade e amizade então demonstradas. Obrigada a ambas por terem partilhado
iii
À Prof. Doutora Octávia Monteiro Gil, do Instituto Tecnológico e Nuclear (ITN), pelo apoio prestado na elaboração da técnica de citogenética. Agradeço também a sua disponibilidade e simpatia.
Ao Centro de Investigação de Genética Médica Humana (CIGMH/FCM-UNL), por me ter recebido em determinadas etapas deste trabalho, nomeadamente na fase de processamento do DNA. A todos os membros da equipa, em especial à Dra. Marta Pingarilho o meu sentido agradecimento pelo apoio prestado.
Desejo também agradecer à Prof. Doutora Isabel Barahona e à Prof. Doutora Ana Clara Ribeiro, coordenadoras do meu trabalho de laboratório no Centro de Investigação Interdisciplinar Egas Moniz (CiiEM-ISCSEM), por permitirem a minha ausência e perceberem o quanto era importante para mim a realização deste projecto. Obrigada pela formação científica prestada ao longo dos últimos anos e apoio nesta etapa. Gostaria ainda de agradecer à Prof. Doutora Isabel Barahona e à Prof. Doutora Madalena Oom, na qualidade de vossa assistente no ISCSEM, as particulares alturas em que facilitaram as minhas funções de docência, contribuindo assim para uma maior disponibilidade da minha parte para este trabalho.
Ao Prof. Doutor Martins dos Santos, presidente da Cooperativa de Ensino Superior Egas Moniz e à direcção do ISCSEM, o meu agradecimento por todo o apoio prestado.
A todos os meus colegas do ISCSEM obrigada pela troca de ideias, apoio e amizade. Aos meus amigos, agradeço o incentivo, confiança e os conselhos quando eu mais precisava. A vossa amizade deu-me a estabilidade necessária para desenvolver este trabalho.
Aos meus pais, irmão e avós agradeço pelo amor e apoio incondicional, a sensatez com que sempre me ajudaram. A ti, pai dedico esta tese. Obrigada pelos valores que me incutiste.
Finalmente, agradeço ao Pedro por partilhar as minhas tristezas e alegrias no decorrer deste trabalho. Obrigada pelo carinho, pela tua tolerância e compreensão nos momentos em que estive ausente.
iv
Nos últimos anos, têm sido observadas quantidades relevantes de acrilamida (AA) num grande número de alimentos ricos em amido após processamento térmico (ex. batatas fritas). Estes dados recentes sugerem que o consumo oral de AA é um factor de risco adicional para o cancro. Neste contexto, a AA tem sido apontada como um potencial agente cancerígeno para o tecido mamário, uma vez que em estudos efectuados com roedores foram observados tumores benignos e malignos da glândula mamária. Um número considerável de evidências apontam para que o metabolito glicidamida (GA), um epóxido gerado pela biotransformação presumivelmente via CYP2E1, desempenhe um papel central na cancerigénese associada à exposição a AA. O trabalho aqui descrito mostra os efeitos citotóxicos e genotóxicos induzidos pela GA em células humanas MCF10A. Estas células mimetizam as células mamárias ductais e têm sido amplamente adoptadas como modelo celular de referência de células de mama não tumorais. As metodologias utilizadas neste trabalho incluem a avaliação da viabilidade celular utilizando o ensaio de redução do MTT, a geração de espécies reactivas de oxigénio (ROS), utilizando o ensaio da dihidrorodamina 123 e a avaliação de genotoxicidade utilizando o teste do micronúcleo em células MCF10A com a citocinese bloqueada.
A GA induziu um decréscimo dependente da concentração da viabilidade celular em células MCF10A. A pré-incubação com butionina sulfoximina (BSO), um inibidor da síntese de glutationo reduzido (GSH), provocou um marcado decréscimo na viabilidade das células tratadas com 1,0 mM de GA (P<0,01), enquanto a incubação concomitante de GSH com GA (3,0 mM) apresentou um ligeiro aumento da viabilidade celular. A co-incubação com superóxido dismutase peguilada (SOD-PEG), catalase peguilada (CAT-PEG), ou um antioxidante catalítico sintético (SOD mimético) não mostrou alterar a viabilidade celular das células MCF10A expostas à GA. Adicionalmente, o tratamento com diferentes concentrações de GA não aumentou os níveis de ROS nas células MCF10A. A exposição de células MCF10A à GA levou a um aumento na frequência de células binucleadas micronucleadas (%MNBN), especialmente para a concentração 1,0 mM (~2 vezes, P <0,01). Para a maior concentração de GA testada (2,0 mM) foi observada uma redução forte da % de células binucleadas, mostrando uma interferência importante ao nível da divisão celular. Em resumo, estes resultados mostram que a GA
v
mecanismo relevante para os efeitos celulares induzidos pela GA em células MCF10A.
Palavras-chave
vi
In the last years, significant amounts of acrylamide (AA) have been confirmed in a wide number of starchy cooked foods (e.g. fried chips). These new data suggest that the oral consumption of AA is an additional risk factor for cancer. In this context, AA has been pointed out as a suspected human breast carcinogen since in rodent studies both benign and malignant mammary gland tumors have been observed. A considerable number of findings strongly suggest that the reactive metabolite glycidamide (GA), an epoxide generated presumably by CYP2E1 biotransformation, plays a central role in AA carcinogenesis. The work reported here addresses GA-induced cytotoxic and genotoxic effects in human MCF10A cells. These cells mimic the mammary ductal cells and have been widely adopted as the reference cell model of non-malignant breast cells. The methodologies used herein included the evaluation of cell viability using the MTT reduction assay, the generation of reactive oxygen species (ROS) using the dihydrorhodamine 123 assay and the assessment of genotoxicity using the cytokinesis-blocked micronucleus assay.
GA induced dose-dependent decrease in MCF10A cell viability. Pre-incubation with buthionine sulfoximine (BSO), a reduced glutathione (GSH) synthesis inhibitor, markedly decreased cell viability of 1 mM GA-treated cells (P<0.01), whereas the concomitant incubation of GSH with GA (3.0 mM) mildly increased cell viability. The incubation with either pegylated superoxide dismutase (SOD-PEG), or pegylated catalase (CAT-PEG) or a synthetic catalytic antioxidant (SOD mimetic) did not modify cell viability of GA exposed cells. Additionally, treatment with different GA concentrations did not increase the levels of ROS in MCF10A cells. The exposure of MCF10A cells to GA led to an increase in the frequency of micronucleated binucleated (%MNBN) cells, especially for 1.0 mM concentration (~2 fold, P<0.01). For the higher concentration tested (2.0 mM), a strong reduction of the % of binucleated cells was observed, showing an important impairment at the cell division level. In summary, these results show that GA is cytotoxic and genotoxic for MCF10A cells, GSH being important for the mitigation of GA toxicity. Moreover, ROS generation does not seem to be a relevant mechanism for GA-induced cellular effects in MCF10A cells.
vii
- 1 -
Índice
Índice ____________________________________________________________________ - 1 - Lista de Abreviaturas _______________________________________________________ - 3 - Lista de Figuras ____________________________________________________________ - 4 - Lista de Tabelas ____________________________________________________________ - 5 - 1. Introdução _____________________________________________________________ - 6 -1.1. A acrilamida: fontes de exposição ______________________________________ - 7 - 1.2. Acrilamida nos alimentos: implicações _________________________________ - 10 -
1.2.1. Mecanismos de formação _________________________________________ - 10 - 1.2.2. Níveis de acrilamida nos alimentos __________________________________ - 13 - 1.2.3. Níveis de exposição ______________________________________________ - 15 -
1.3. Toxicocinética _____________________________________________________ - 17 - 1.4. Toxicodinâmica ____________________________________________________ - 21 -
1.4.1. Espectro de efeitos tóxicos ________________________________________ - 24 -
1.5. Acrilamida como provável cancerígeno humano _________________________ - 27 -
1.5.1. Genotoxicidade _________________________________________________ - 27 - 1.5.1.1. Mutações genéticas em células procariotas ________________________ - 27 - 1.5.1.2. Mutações genéticas em células de mamífero _______________________ - 28 - 1.5.1.3. Ensaios citogenéticos _________________________________________ - 30 - 1.5.1.4. Formação de aductos de DNA __________________________________ - 32 - 1.5.2. Cancerigénese em modelo animal ___________________________________ - 34 - 1.5.2.1. Tumores da glândula mamária em ratos __________________________ - 39 - 1.5.3. Estudos epidemiológicos __________________________________________ - 41 - 1.5.3.1. Evidências de tumores do tecido mamário em humanos ______________ - 45 -
1.6. Modo de acção na cancerigénese ______________________________________ - 51 -
1.6.1. Hipótese de mutagenicidade _______________________________________ - 51 - 1.6.2. Hipótese de desregulação dos níveis hormonais e sinalização _____________ - 53 -
1.7. Posição actual ______________________________________________________ - 55 - 2. Objectivos _____________________________________________________________ - 57 - 3. Materiais e Métodos _____________________________________________________ - 59 - 3.1. Materiais _________________________________________________________ - 60 - 3.1.1. Reagentes ______________________________________________________ - 60 - 3.1.2. Linha celular ___________________________________________________ - 61 - 3.2. Métodos __________________________________________________________ - 62 -
3.2.1. Cultura de células MCF10A _______________________________________ - 62 - 3.2.2. Ensaio de redução do MTT ________________________________________ - 63 - 3.2.2.1. Determinação da viabilidade celular _____________________________ - 64 - 3.2.2.2. Modulação de glutationo na citotoxicidade da glicidamida ____________ - 64 - 3.2.2.3. Efeitos de antioxidantes na citotoxicidade da glicidamida ____________ - 65 - 3.2.3. Formação de espécies reactivas de oxigénio ___________________________ - 67 -
- 2 -
3.2.4. Ensaio do micronúcleo em células com a citocinese bloqueada ____________ - 68 - 3.2.5. Determinação de aductos específicos de DNA _________________________ - 70 - 3.2.5.1. Exposição química e extracção de DNA __________________________ - 70 - 3.2.5.2. Quantificação do DNA _______________________________________ - 71 - 3.2.5.3. Quantificação dos aductos de DNA ______________________________ - 71 - 3.2.6. Análise estatística _______________________________________________ - 71 -
4. Resultados e Discussão __________________________________________________ - 73 -
4.1. Avaliação do potencial citotóxico da GA em MCF10A ____________________ - 74 -
4.1.1. Citotoxicidade da glicidamida ______________________________________ - 75 -
4.2. Efeito de moduladores do GSH na citotoxicidade da GA __________________ - 78 -
4.2.1. Efeito da deplecção de glutationo ___________________________________ - 78 - 4.2.2. Efeito da incubação com glutationo__________________________________ - 80 -
4.3. Potencial envolvimento de um mecanismo de stress oxidativo ______________ - 83 -
4.3.1. Efeito de antioxidantes na citotoxicidade da glicidamida _________________ - 83 - 4.3.2. Quantificação de espécies reactivas de oxigénio ________________________ - 88 -
4.4. Avaliação do potencial genotóxico da GA em MCF10A ___________________ - 90 -
4.4.1. Avaliação da indução de MN ______________________________________ - 91 - 4.4.2. Formação de aductos específicos de DNA ____________________________ - 95 -
5. Conclusões e Perspectivas Futuras _________________________________________ - 96 - Referências Bibliográficas _________________________________________________ - 100 -
- 3 -
Lista de Abreviaturas
8-OHdG 8-hidroxidesoxiguanosina
AA Acrilamida
BMD Benchmark Dose
BMDL Benchmark Dose Lower Confidence Limit
BN Binucleada
BSO Butionina Sulfoximina
CAs Aberrações Cromossómicas
CAT Catalase
CAT-PEG Catalase Peguilada
Cit-B Citocalasina B
CYP2E1 Citocromo P450 2E1
DHR123 Dihidrorodamina 123
DMEM/F12 Dulbecco's Modified Eagle Medium nutrient mixture F-12 Ham
DNA Ácido Desoxirribonucleico
EDTA Ácido Etilenodiaminotetracético
EGF Factor de Crescimento Epidérmico Humano
EH Epóxido Hidrolase
ER Receptor de Estrogénio
GA Glicidamida
GSH Glutationo reduzido (γ-L-Glutamil-L-cisteinil-glicina)
GST Glutationo-S-Transferase
Hb Hemoglobina
HPRT Hipoxantina Guanina Fosforibosiltransferase
IC50 Concentração inibitória de 50% do crescimento celular
ip Intraperitoneal
LOAEL Lowest Observed Adverse Effect Level
MCF10A Células epiteliais de tecido mamário humano não tumorais
MN Micronúcleo
MNCB Micronúcleo em células com a citocinese bloqueada MTT Brometo de 3-(4,5-dimetil-tiazol-2-il)-2,5 difenil-tetrazólio
NADH Nicotinamida Adenina Dinucleótido Reduzido
NADPH Nicotinamida Adenina Dinucleótido Fosfato Reduzido
NDI Índice de Divisão Nuclear
NOAEL No Observed Adverse Effect Level
pc Peso Corporal
PEG Poli (Etileno Glicol)
PR Receptor de Progesterona
QFA Questionário de Frequência Alimentar
ROS Espécies Reactivas de Oxigénio
SCE Trocas entre Cromátides-Irmãs
SNC Sistema Nervoso Central
SOD Superóxido Dismutase
SODm Superóxido Dismutase Mimético
SOD-PEG Superóxido Dismutase Peguilada
TBHP Tert - Butil-Hidroperóxido
TE Tris - Ácido Etilenodiaminotetracético
TK Timidina Cinase
UDS Síntese de DNA não programada
UE União Europeia
- 4 -
Lista de Figuras
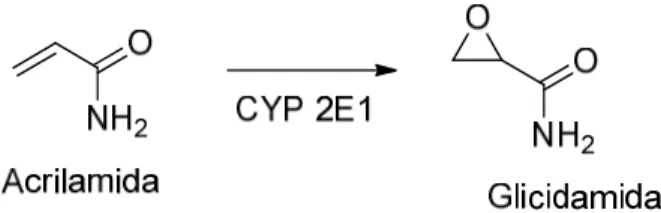
Figura 1. Biotransformação da acrilamida em glicidamida mediada pelo citocromo P450 2E1. ... 9
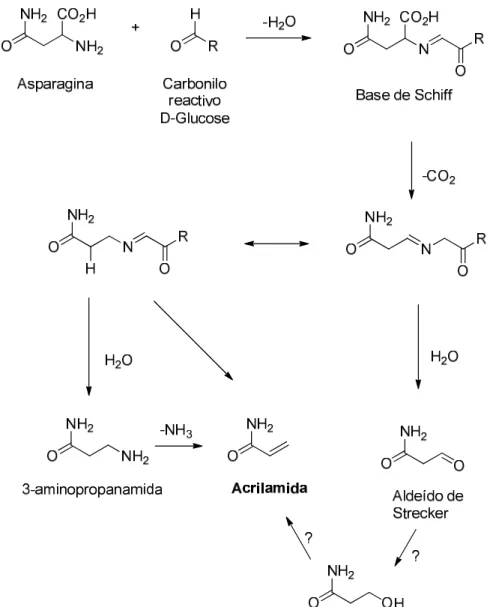
-Figura 2. Mecanismo proposto para a formação de acrilamida nos alimentos processados termicamente. ... 11
Figura 3. Via metabólica proposta para a AA nos mamíferos... ... 18
Figura 4. Estrutura química dos principais aductos de DNA da acrilamida e da glicidamida. ... 23
Figura 5. Fotografias de microscopia óptica de uma cultura de células MCF10A. ... 61
Figura 6. Fluxograma simplificado do ensaio de redução do MTT.. ... 66
-Figura 7. Fluxograma simplificado do método de determinação de espécies reactivas de oxigénio intracelulares. ... 68
-Figura 8. Fluxograma simplificado do método padrão do ensaio do micronúcleo com citocinese bloqueada. ... 69
-Figura 9. Avaliação da citotoxicidade da glicidamida (GA, 0,1 a 4,0 mM; 24h de incubação) em células MCF10A, usando o ensaio de redução do MTT. ... 76
-Figura 10. Efeito do BSO (100 µM; pré-tratamento de 24h) na potenciação do efeito citotóxico da glicidamida (GA 1 mM; 24h de incubação), medido através do decréscimo da viabilidade celular de células MCF10A. ... 79
-Figura 11. Efeito do GSH (1 mM; co-incubação de 24h) na diminuição do efeito citotóxico da glicidamida, medido através do aumento da viabilidade celular de células MCF10A. ... 82
-Figura 12. Efeito de antioxidantes na viabilidade celular induzida pela glicidamida (GA 1 mM; 24h de co-incubação) em células MCF10A, determinado pela redução do MTT e apresentado como percentagem relativa ao controlo de células não expostas... 86
-Figura 13. Efeito da glicidamida nos níveis intracelulares de ROS em células MCF10A, avaliado por oxidação de DHR123. ... 89
Figura 14. Efeito da glicidamida na proliferação celular/divisão celular das células MCF10A. ... 93
-Figura 15. Genotoxicidade da glicidamida nas células MCF10A, expressa em ‰ de células binucleadas micronucleadas (‰ MNBN). ... 94
-- 5 --
Lista de Tabelas
Tabela I. Níveis de acrilamida em vários grupos de alimentos, reportados pelos Estados Membros da UE e Noruega, referentes ao ano 2007 e 2008. ... - 14 - Tabela II. Incidência de tumores nos bioensaios crónicos com ratos Fisher 344, expostos à
acrilamida na água de abastecimento, durante 2 anos.. ... - 35 - Tabela III. Resultados dos estudos de tumorigénese da acrilamida no ratinho. . ... 38 -Tabela IV. Resumo dos pontos de partida estimados para os conjuntos de dados de tumores em
ratos, inclusive tumores mamários, usando como dose a acrilamida e a glicidamida. ... 40 -Tabela V. Estudos epidemiológicos de avaliação do risco de cancro por ingestão de acrilamida
através da dieta. ... - 43 - Tabela VI. Estudos epidemiológicos de ingestão de acrilamida através da dieta e risco de cancro
de mama. ... 46 Tabela VII. Materiais utilizados no presente trabalho. ... 60
-- 6 --
- 7 -
1.1.
A acrilamida: fontes de exposição
Desde 1950 que a acrilamida (AA) é produzida em larga escala através da hidratação do acrilonitrilo, principalmente para a formação de poliacrilamida. Na forma monomérica, este sólido cristalino, de baixo peso molecular, inodoro e incolor1, participa facilmente em reações de polimerização, cujos produtos constituem a base de uma vasta aplicação industrial e laboratorial. As suas príncipais utilizações são como agente floculante no processo de depuração de água potável e tratamento de águas residuais urbanas e industriais e como agente de controlo de fluxo em operações em poços de petróleo. Por outro lado, é utilizada em engenharia civil, em fundações, como constituinte da argamassa utilizada na construção e reparação de esgotos, túneis e barragens. A AA é ainda utilizada na estabilização de solos, síntese de corantes, produção de materiais de embalagem, co-polímeros para lentes de contacto, cosméticos e em muitos laboratórios de biologia molecular e engenharia genética, na preparação de géis de electroforese (IARC, 1994; WHO, 2003).
Até à decada de 90, foram publicados inúmeros estudos toxicológicos da AA dando a conhecer o seu efeito cancerígeno em modelo animal e o seu efeito neurotóxico em animais e humanos (IARC, 1994). No entanto, continuou a haver uma longa e ampla utilização da AA e só em 1997 ocorreu o primeiro sinal de alarme, como resultado de uma exposição humana ocupacional à AA durante a contrução de um túnel ferroviário no Sudoeste da Suécia (Törnqvist et al., 2000). O alerta foi dado pelo aparecimento de um elevado número de peixes mortos e vacas paralisadas num rebanho que consumia água a partir de um riacho nas proximidades da construção. A detecção de elevados níveis de aductos de AA com a hemoglobina (AA-Hb) nos animais contaminados, associados aos níveis incrivelmente elevados de AA no riacho contaminado, não só esclareceu a origem do problema como incentivou o urgente estudo dos mais de 200 trabalhadores e residentes da área expostos a AA. Os resultados mostraram que grande parte dos trabalhadores tinha níveis elevados de aductos de exposição a AA. Neste episódio, 50 trabalhadores alegaram sintomas de neurotoxicidade e para 23 deles houve forte evidência de comprometimento do sistema nervoso periférico (Törnqvist et al., 2000; Törnqvist, 2005). Contudo, a avaliação de risco considerou que haveria uma
- 8 -
probabilidade pequena de risco de cancro para os trabalhadores expostos pela via dérmica e inalatória à AA (Törnqvist et al., 2000; Törnqvist, 2005).
A opinião geral durante a década de 90 era de que o homem raramente estava exposto à AA em circunstâncias normais. Acreditava-se que a principal via de exposição humana à AA era de origem ocupacional, centrando as preocupações para este nicho da população. O público em geral foi considerado exposto apenas a pequenas quantidades no consumo de água potável, refinada com poliacrilamida, ou pela inalação do fumo do tabaco, no caso dos fumadores (IARC, 1994). Todavia, alguns estudos na área mostraram níveis de base elevados de aductos de AA-Hb em indivíduos não expostos (grupos de controlo) (Bergmark, 1997) e os autores não encontraram explicação para o resultado, que consideraram inesperado, mas que indicava uma possível fonte de exposição generalizada. Uma parte da explicação apareceu quando Tareke e colaboradores (Tareke et al., 2000) encontraram, em ratos alimentados com dieta animal padrão frita, um aumento nos níveis de aductos de AA-Hb, quando comparado com o grupo controlo (alimentado com dieta não frita). Esta observação no inicio do ano 2000 foi amplamente ignorada. No entanto, dois anos mais tarde, os mesmos investigadores da Universidade de Estocolmo comunicaram a descoberta de níveis relativamente elevados de AA em diversos alimentos, ricos em amido e sujeitos a tratamento térmico (Tareke et al., 2002). Esta mensagem eclodiu na comunicação social e causou preocupação entre a população em geral e na comunidade científica sobre os possíveis riscos para a saúde humana.
A descoberta inesperada de que os seres humanos possam estar regularmente expostos a doses relativamente elevadas de AA através do consumo normal de alimentos processados foi, e é, o resultado de uma investigação sistemática e desenvolvimentos relevantes nas metodologias ao longo de décadas, aliadas a uma cadeia de coincidências. Este facto deu início a um intenso levantamento analítico dos produtos alimentares e estudos mecanísticos para identificar as condições que levam à formação de AA. A Swedish National Food Administration (SNFA) foi o primeiro órgão que realizou um estudo sobre a determinação de AA em produtos disponíveis no mercado, confirmando a sua presença em diferentes níveis, em muitos alimentos processados termicamente (FAO/WHO, 2002). Desde então, várias entidades competentes, a nível mundial, têm avaliado o teor de AA nos alimentos e todas elas reportaram a presença de AA em produtos e em níveis semelhantes aos inicialmente determinados no país nórdico (FAO/WHO, 2005; EFSA, 2010; EPA, 2010).
- 9 -
Desde 2002, as investigações sobre a presença de AA nos alimentos e as possíveis consequências na saúde humana têm-se sucedido de forma continuada e vários progressos têm sido alcançados (Rice, 2005; Parzefall, 2008). O material publicado desde essa data, só referente ao termo “acrylamide in food”, ascende a mais de 500 artigos científicos2 até à data. Todo o conjunto de conhecimento adquirido é fruto, sem
dúvida, de uma comunidade científica que respondeu positivamente a um problema de proporções mundiais em termos de saúde pública. O enorme esforço dispendido pelos vários grupos de investigação para compreender os mecanismos de acção, as doses relevantes e os seus efeitos tóxicos, teve e tem como objectivo a compreensão global dos riscos associados à exposição humana. Contudo, no que diz respeito à sua toxicidade, o peso das provas para a AA em estudos epidemiológicos e em animais de experiência são aparentemente conflituosos, sendo a avaliação de risco relativa ao consumo de AA uma das mais controversas actualmente. O risco associado à ingestão de AA não está totalmente esclarecido e vários estudos continuam em curso (EFSA, 2008; EPA, 2010).
Indubitavelmente, a constatação de um alto teor do composto tóxico AA em alimentos abriu áreas para novas pesquisas apelando para um novo pensamento em vários domínios. A sua ocorrência, activação metabólica, genotoxicidade e risco de cancro humano são os tópicos actuais em discussão.
Neste âmbito, há varias décadas que se sabe que a AA é biotransformada por epoxidação em glicidamida (GA) (Figura 1). No entanto, foi em 1993 que surgiu a evidência da formação de GA nos seres humanos, mais precisamente num grupo de trabalhadores expostos a níveis relativamente elevados de AA (Bergmark et al., 1993).
Figura 1. Biotransformação da acrilamida em glicidamida mediada pelo citocromo P450 2E1.
2De acordo com as publicações disponíveis no NCBI- National Center for Biotechnology
- 10 -
A GA e o seu composto parental são electrófilos muito solúveis em água. Ambos reagem com centros nucleófilos nas macromoléculas biológicas, em adições de Michael (Calleman, 1996), originando aductos, inclusive com a Hb (Friedman, 2003). Comparada com a GA, a AA tem uma reactividade electrofílica relativamente superior com os grupos sulfidrilo (SH) (Bergmark et al., 1993) e baixa reactividade com o DNA (Besaratinia and Pfeifer, 2005). A GA, porém, tem demonstrado formar aductos com as bases púricas do DNA em quantidades apreciáveis (Gamboa da Costa et al., 2003; Doerge et al., 2005a). Na verdade, a mutagenicidade associada à AA é maioritariamente atribuída à sua conversão em GA (Besaratinia and Pfeifer, 2005; Exon, 2006), focando grande parte do interesse toxicológico deste composto no seu metabolito e tornando-o alvo de um número apreciável de estudos. Contudo, o papel da GA na toxicidade da AA não está totalmente esclarecido e continua a ser merecedor de atenção por parte da comunidade científica.
1.2.
Acrilamida nos alimentos: implicações
1.2.1. Mecanismos de formação
Diversos autores demonstraram que a formação de AA em alimentos envolve a reacção de Maillard, também conhecida como reacção de escurecimento, entre aminoácidos e açúcares redutores (Mottram et al., 2002; Stadler et al., 2002). Especificamente, a reacção ocorre entre o grupo carbonilo da glucose e o grupo amina livre dos aminoácidos asparagina > glutamina > cisteína > metionina, na ordem de eficiência apresentada, desde que estes elementos essenciais para a formação de AA sejam aquecidos acima de 120 ºC (Stadler et al., 2002).
De um modo geral, o principal mecanismo proposto para a formação da AA compreende a formação de uma base de Schiff a partir da reacção entre o aminoácido e a glucose. A descarboxilação dessa base de Schiff dá origem a um composto intermediário instável que pode ser hidrolisado a 3-aminopropanamida, a qual por perda de amónia produz AA. Alternativamente, o composto intermediário pode formar AA através da clivagem da ligação CN dando origem a AA, como apresentado na Figura 2 (Mottram et al., 2002; Friedman, 2003; Heatox, 2007). A base de Schiff, por sua vez,
- 11 -
também pode sofrer rearranjos e originar os designados produtos da reacção de Maillard responsáveis por muitos dos sabores e cores gerados durante o cozimento e fritura dos alimentos ou sofrer reacções de degradação de Strecker, em que o aminoácido é descarboxilado e desaminado para formar um aldeído. Este último pensa-se que também se poderá transformar em AA (Figura 2) (Mottram et al., 2002).
Assim, a AA é produzida naturalmente em alguns alimentos processados a altas temperaturas e os teores variam consoante a cinética das reacções, que têm vindo a ser extensivamente estudadas, mostrando uma correlação com o escurecimento do produto (Heatox, 2007).
Figura 2. Mecanismo proposto para a formação de acrilamida nos alimentos processados termicamente. Adaptado de Mottram et al. (2007) e Heatox (2007).
- 12 -
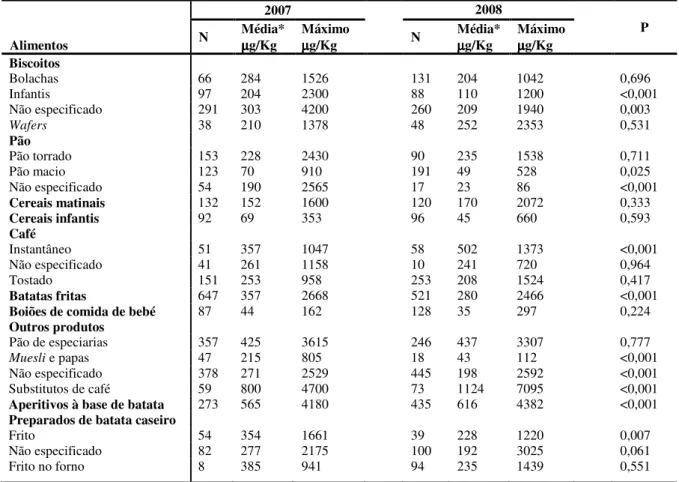
As variações observadas nos níveis de AA em diversas categorias de alimentos, bem como em diferentes marcas dentro da mesma categoria (Svensson et al., 2003) parecem resultar não só dos níveis de percusores presentes, mas também de variações nas condições de processamento (por exemplo, temperatura, tempo de preparação, natureza dos óleos de fritura e natureza da matriz alimentar). Os conhecimentos adquiridos em relação aos factores que influenciam a formação de AA nos alimentos têm sido úteis no desenvolvimento de estratégias de diminuição do potencial de formação deste contaminante alimentar. Ao longo destes anos, os produtores comerciais, com a ajuda das entidades reguladoras, tentaram adaptar os seus processos de produção com o intuito de reduzir a formação de AA. No entanto, o problema continua a consistir no facto de, apesar de já existirem formas de reduzir a quantidade de AA, não existir forma de a eliminar ou reduzir para níveis insignificantes (Heatox, 2007). Aliás, a última compilação de dados referentes ao ano de 2008, reportada no presente ano (EFSA, 2010), apresenta 3461 resultados (num total de 22 Estados Membros da União Europeia e a Noruega) nos quais se verifica que a categoria de produtos “café instantâneo” e “substitutos de café” apresentam níveis significativamente superiores de AA nos dados de 2008 comparado com os dados de 2007 (Tabela I). Por outro lado, grande parte das outras categorias apresenta níveis significativamente mais baixos quando comparado com dados de 2007, mas não inexistentes. Supõe-se que a aplicação das estratégias de redução apenas foi eficaz num número limitado de grupos de alimentos (EFSA, 2010). O teor de AA nos alimentos é desenvolvido no ponto seguinte (1.2.2).
Para concluir, a AA não é o único composto formado durante o processamento dos alimentos. Os mesmos investigadores que deliberaram sobre o mecanismo de formação de AA nos alimentos, criaram uma base de dados com mais de 800 compostos formados pela acção da temperatura nos alimentos, dos quais cerca de 50 poderão ser agentes potencialmente cancerígenos (Heatox, 2007; Ferguson and Philpott, 2008). Como exemplos, destacam-se os compostos N-nitroso, hidrocarbonetos aromáticos policíclicos como o benzo(a)pireno, e as aminas heterocíclicas, incluindo a 2-amino-1-metil-6-fenilimidazo[4,5-b]piridina. Todos estes compostos são reactivos com o DNA e dão origem a espectros distintos de mutações por substituição de pares de bases e alterações cromossómicas estruturais (Ferguson and Philpott, 2008). Este facto poderá ser determinante nas vias de acção tóxica da AA, alertando para o estudo da AA quer na sua componente isolada, quer na componente conjunta.
- 13 - 1.2.2. Níveis de acrilamida nos alimentos
Os alimentos ricos em percursores da AA são em grande parte derivados de fontes vegetais como batatas e cereais (cevada, trigo, arroz), mas aparentemente não de alimentos de origem animal, como aves, carnes e peixes (FAO/WHO, 2005). Os dados analíticos mostraram que os principais itens alimentares, com maior contribuição na dieta ocidental, para a exposição total à AA foram as batatas fritas (6-46%)3, café
(13-39%), biscoitos e pastelaria em geral (10-20%) e o pão e tostas (10-30%). Os outros itens alimentares contribuiam com menos de 10% para a exposição total à AA (FAO/WHO, 2005).
Os últimos dados do teor de AA nos alimentos, reportados pelos Estados Membros da União Europeia (UE) e pela Noruega, referentes aos anos de 2007 e 2008 e compilados pela European Food Safety Authority (EFSA), são apresentados na Tabela I. Portugal foi um dos países que não disponibilizou resultados dos níveis de AA nos itens alimentares, não contribuindo assim para o primeiro e segundo de uma série de três relatórios da EFSA (sobre os anos de 2007, 2008 e 2009, respectivamente), que auxiliarão a Comissão Europeia e os Estados Membros a determinar se as medidas levadas a cabo voluntariamente pela indústria alimentar para reduzir os níveis de AA nos alimentos são eficazes.
Segundo os dados apresentados na Tabela I (EFSA, 2010), nos resultados reportados nas amostras de alimentos de 2008, os níveis médios de AA mais elevados foram observados no grupo de alimentos “substitutos de café”, que inclui bebidas semelhantes ao café à base de cereais como cevada ou chicória. Este grupo também revelou conter o valor máximo de AA, em ambos os anos de amostragem, entre todos os géneros alimentícios testados (4700 µg/Kg em 2007 e 7095 µg/Kg em 2008). Outros géneros alimentícios que apresentaram níveis de AA elevados, nos dados mais recentes, correspondem aos aperitivos à base de batata e café instantâneo. Por outro lado, dos 22 grupos de alimentos estudados, os valores médios mais baixos foram encontrados em produtos de panificação (23 µg/Kg) e nos boiões de comida para bebé (44 µg/Kg), respectivamente para as amostras recolhidas em 2008 e 2007.
- 14 -
Tabela I. Níveis de acrilamida em vários grupos de alimentos, reportados pelos Estados Membros da UE e Noruega, referentes ao ano 2007 e 2008. Adaptado de EFSA (2010).
Alimentos 2007 2008 P N Média* µ µµ µg/Kg Máximo µ µ µ µg/Kg N Média* µ µµ µg/Kg Máximo µ µ µ µg/Kg Biscoitos Bolachas 66 284 1526 131 204 1042 0,696 Infantis 97 204 2300 88 110 1200 <0,001 Não especificado 291 303 4200 260 209 1940 0,003 Wafers 38 210 1378 48 252 2353 0,531 Pão Pão torrado 153 228 2430 90 235 1538 0,711 Pão macio 123 70 910 191 49 528 0,025 Não especificado 54 190 2565 17 23 86 <0,001 Cereais matinais 132 152 1600 120 170 2072 0,333 Cereais infantis 92 69 353 96 45 660 0,593 Café Instantâneo 51 357 1047 58 502 1373 <0,001 Não especificado 41 261 1158 10 241 720 0,964 Tostado 151 253 958 253 208 1524 0,417 Batatas fritas 647 357 2668 521 280 2466 <0,001
Boiões de comida de bebé 87 44 162 128 35 297 0,224
Outros produtos
Pão de especiarias 357 425 3615 246 437 3307 0,777
Muesli e papas 47 215 805 18 43 112 <0,001
Não especificado 378 271 2529 445 198 2592 <0,001
Substitutos de café 59 800 4700 73 1124 7095 <0,001
Aperitivos à base de batata 273 565 4180 435 616 4382 <0,001
Preparados de batata caseiro
Frito 54 354 1661 39 228 1220 0,007
Não especificado 82 277 2175 100 192 3025 0,061
Frito no forno 8 385 941 94 235 1439 0,551
N – tamanho da amostra; P – significância da análise de variância entre sub-grupos de alimentos, quando comparado os resultados entre 2007 e 2008. * Os valores médios apresentados correspondem ao limite superior de ingestão.
Quando comparados os resultados de 2008 com os resultados recolhidos em 2007, os níveis de AA reportados parecem ter sido mais baixos em 2008 do que em 2007 de um modo global. No entanto, esta redução não ocorreu em todos os grupos de alimentos, como referido no ponto anterior. Alguns apresentaram níveis mais elevados de AA em 2008, nomeadamente os géneros alimentícios observados com níveis mais elevados em todo o grupo analisado. Contudo, foi verificada uma diminuição significativa para os grupos de preparados de batata caseiros, batata frita, produtos de panificação, papas e
muesli e em produtos não especificados. Os outros 50% de produtos analisados, entre
eles os cereais e os biscoitos, não apresentaram diferenças significativas entre os dados disponibilizados nos dois anos (EFSA, 2010).
Em particular, os dados publicados sobre os teores de AA na alimentação portuguesa são escassos, sendo que os hábitos alimentares dos portugueses, nomeadamente a característica dieta mediterrânica ou formas tradicionais de preparação dos alimentos,
- 15 -
distintos dos utilizados na grande maioria dos países europeus, justificam um estudo detalhado para a realidade portuguesa.
1.2.3. Níveis de exposição
O consumo médio diário de AA foi estimado em cerca de 0,5-1,0 µg/Kg peso corporal (pc) nos adultos e até duas vezes mais nas crianças com idade compreendida entre os 9 os 13 anos, por consumo de uma dieta normal ocidental (Dybing and Sanner, 2003), particularmente avaliada na Noruega. Com base na população norueguesa, a maior fonte de ingestão de AA para as crianças foram as batatas fritas, mas os biscoitos de manteiga e outros doces também foram fontes importantes. Estes produtos contribuiram em aproximadamente 55-65% da ingestão média total das crianças entre os 9 e os 13 anos de idade. Nos adultos, as fontes mais relevantes foram o café, as batatas fritas e o pão. Este último, embora contenha pequenas quantidades de AA é consumido diariamente em montantes relativamente elevados na Noruega (Dybing and Sanner, 2003).
Um estudo recente mostrou pela primeira vez variabilidade nos dados de exposição à AA adquiridos com a mesma metodologia analítica, entre grupos de 9 países. Neste estudo, os valores máximos observados no grupo britânico, holandês e grego foram maiores do que os observados nos restantes grupos. A explicação dos autores prende-se com o facto de a dieta destes países ser mais rica em batata (Vesper et al., 2008). Porém, os dados oficiais de ingestão mais recentes da Organização Mundial de Saúde (World Health Organization - WHO), mostraram que o consumo médio de AA podia variar a nivel nacional de 0,3 a 2 µg/Kg peso corporal (pc)/dia, podendo mesmo chegar a 5,1 µg/Kg pc/dia, no percentil 99 (FAO/WHO, 2005). Com base nos dados disponíveis, as crianças apresentavam valores de ingestão de AA duas a três vezes superiores aos encontrados em consumidores adultos, quando este resultado foi expresso em peso corporal. Desta forma, o Joint FAO/WHO Committee on Food
Additives (JECFA) concluiu que uma ingestão de 1 µg/Kg pc/dia de AA poderia ser
utilizada para representar a média da população em geral e que uma ingestão de 4 µg/Kg pc/dia poderia ser utilizada para representar os grandes consumidores, incluindo as crianças, nestas estimativas de consumo médio e elevado (FAO/WHO, 2005).
- 16 -
A faixa etária das crianças e adolescentes é, sem dúvida, um factor a ter em consideração, sobretudo porque o consumo elevado de produtos denominados snacks, bem como o menor peso destes indivíduos, origina valores estimados de ingestão significativamente superiores e caracteriza-os como um grupo de risco (Dybing and Sanner, 2003). Adicionalmente, os seres humanos em geral estão expostos à AA através de outras vias de exposição. Para além da formação de AA nos alimentos, pequenas quantidades de polímeros de AA são usados em sistemas de água, no tratamento antes da distribuição à população em geral. Alguma, embora pequena, quantidade de monómero pode estar presente na água consumida da rede pública4 (WHO, 2003). Além disso, a AA é um dos componentes do fumo do cigarro. O conteúdo de AA no fumo do cigarro foi estimado ser de cerca de 1,1-2,34 µg/cigarro (Smith et al., 2000). Boettcher
et al. (2005), mediram os metabolitos da AA na urina humana e relataram níveis médios
em indivíduos fumadores cerca de quatro vezes maiores do que em não fumadores, indicando que o fumo do cigarro é claramente uma fonte de exposição à AA. Como consequência, a maioria dos seres humanos está exposta repetidamente a baixos níveis de AA na sua dieta e noutras fontes, tais como o fumo do tabaco inalado.
Em resumo, até 2002, a AA foi considerada essencialmente como um tóxico industrial ou ocupacional, e as vias de exposição consideradas foram sobretudo a absorção dérmica e a inalação de aerossóis no local de trabalho. Os novos dados sugerem o consumo oral de AA como um elemento chave na avaliação do risco global. Aliás, actualmente pensa-se que a dieta é a principal fonte de exposição à AA entre os indivíduos não fumadores (Boettcher et al., 2006a), mas a magnitude do risco que a sua ingestão, através dos alimentos, representa para a saúde humana não está totalmente compreendida (EFSA, 2008). Apesar da presença deste composto formado pela acção da temperatura nos alimentos ser uma realidade, os riscos de toxicidade aguda relacionados com a presença de contaminantes deste tipo nos alimentos são mínimos. Os níveis de AA presentes numa alimentação variada e equilibrada são muito pequenos (EFSA, 2008; EFSA, 2010). No entanto, em termos de toxicidade crónica, torna-se mais complicada a avaliação dos seus efeitos biológicos para o consumidor, principalmente quando existe uma elevada possibilidade de acção conjunta, ou seja de mistura de contaminantes alimentares no organismo (Jägerstad and Skog, 2005; Ferguson and
4A União Europeia estabeleceu, através da Directiva EU 98/83, 0,1 µg/L como valor máximo para a
- 17 -
Philpott, 2008) e porque a toxicidade está directamente relacionada com a cinética de degradação destes compostos no organismo (EFSA, 2008; Gargas et al., 2009).
1.3.
Toxicocinética
Actualmente, está definido que a maioria da AA ingerida é absorvida pelo tracto grastrointestinal, enquanto a absorção por inalação ou exposição dérmica é muito menos eficiente (Fennell et al., 2006; Fuhr et al., 2006). Experiências em ratos, cães e porcos demonstraram que, para além de ser rapidamente absorvida, também é rapidamente distribuída por todos os tecidos (Parzefall, 2008), incluindo o tecido mamário (Doerge
et al., 2005b; Doerge et al., 2005c). A AA foi também encontrada no leite materno
humano e foi demonstrado que também penetra através da placenta humana (Sörgel et
al., 2000).
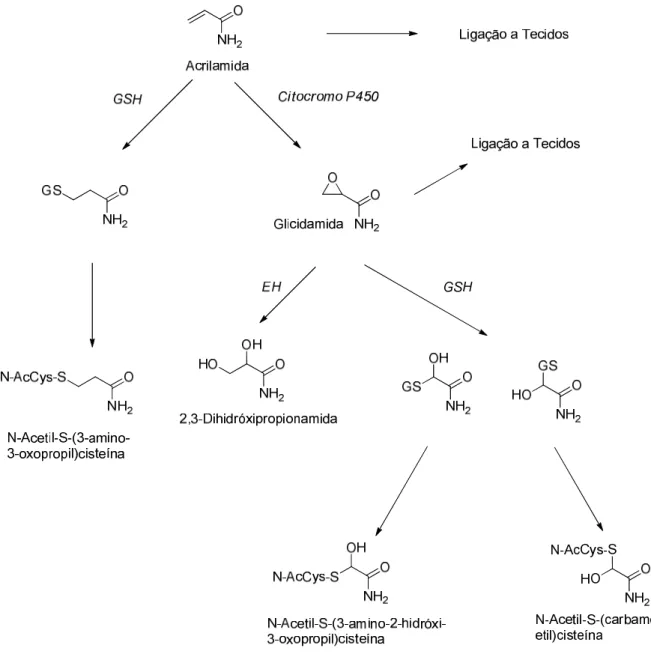
O metabolismo da AA tem sido extensivamente estudado em roedores e seres humanos e inclui reacções de fase I e de fase II de metabolização (Gargas et al., 2009), esquematizadas na Figura 3.
Tanto em roedores como nos seres humanos, uma fracção da AA ingerida é convertida por vias oxidativas, catalisadas presumivelmente pelo citocromo P450 2E1 (CYP2E1), em GA (fase I do metabolismo), um derivado de epóxido que é substancialmente mais reactivo com as macromoléculas que a própria AA (Sumner et
al., 1999; Doroshyenko et al., 2009). Em 1999, um grupo de investigadores (Sumner et al., 1999) estudou o papel do CYP2E1 nos ratinhos, concluindo que é necessário um
CYP2E1 funcional para a conversão in vivo de AA em GA, pelo menos em níveis detectáveis através dos metabolitos urinários. Desde então, vários estudos foram efectuados e o CYP2E1 é hoje classificado como a príncipal enzima responsável pela metabolização de AA em GA em roedores e seres humanos (Ghanayem et al., 2005a; Settels et al., 2008; Doroshyenko et al., 2009).
Na fase II do metabolismo, ambas AA e GA são destoxificadas pela acção da enzima glutationo-S-transferase (GST), a qual catalisa a conjugação com o glutationo (GSH), ou por conjugação espontânea com GSH. Adicionalmente, a GA é inactivada pela epóxi-hidrolase dando origem à gliceramida (2,3-dihidróxipropionamida) (Figura 3) (Calleman, 1996; Fuhr et al., 2006; Kopp and Dekant, 2009).
- 18 -
Figura 3. Via metabólica proposta para a AA nos mamíferos. GSH, glutationo; EH, epóxido hidrolase. Adaptado de Gargas et al. (2009).
Exceptuando as diferenças relacionadas com o metabolismo da AA e seus metabolitos, poucas diferenças qualitativas são esperadas entre humanos e roedores em relação à absorção, distribuição e excreção, em parte porque a AA é muito hidrofílica (Gargas et al., 2009). No entanto, entre os seres humanos, a absorção de AA pelas crianças mostrou ser superior (1,3-1,5 vezes) à dos adultos (Hartmann et al., 2008).
A via urinária é a principal via de excreção dos metabolitos da AA. Após conjugação com o glutationo, a AA e a GA são rapidamente eliminadas na urina como derivados de ácidos mercaptúricos, (3-amino-3-oxopropil)cisteína (AAMA) e N-acetil-S-(3-amino-2-hidroxi-3oxopropil)cisteína (GAMA), respectivamente (Figura 3)
- 19 -
(Boettcher et al., 2006b; Fuhr et al., 2006). A biodisponibilidade absoluta de AA em roedores e seres humanos é dependente do mecanismo de destoxificação e da dose ingerida (Gargas et al., 2009). Fennell e Friedman constataram que ratos tratados com 3 mg/Kg de AA originaram cerca de 59% de metabolitos urinários resultantes da conjugação de AA e GSH (Fennell and Friedman, 2005). Por sua vez, o homem excretou cerca de 30% a mais do que os ratos numa dose equivalente de AA (ou seja, 86%) (Fennell et al., 2005), indicando uma menor biodisponibilidade de AA para reacções de fase I de metabolização. Em contrapartida, os mesmos investigadores demonstraram, na mesma dose administrada, uma recuperação de cerca de 20% de conjugados de GA-GSH na urina dos ratos (Fennell et al., 2005), enquanto as excreções de conjugados de GA-GSH no homem estava abaixo do limite de quantificação (isto é, pelo menos numa ordem de grandeza a menos do que os ratos) (Fennell and Friedman, 2005). Contudo, outros estudos toxicocinéticos em voluntários revelaram excreções de conjugados de GA-GSH quantificáveis (Boettcher et al., 2006b; Fuhr et al., 2006). No homem, foi observada uma recuperação urinária global (AAMA e GAMA) de cerca de 51% de uma dose de 1 mg de AA após 24h (Boettcher et al., 2006b) e de cerca de 60% de uma dose de 0,94 mg de AA após 72h (Fuhr et al., 2006), apontando o AAMA como o principal metabolito urinário da AA (cerca de 50% no total dos produtos excretados), mas não o único. Ambos os estudos, demonstraram que o metabolito urinário GAMA representava cerca de 5-6% da dose de AA administrada (Boettcher et al., 2006b; Fuhr
et al., 2006).
Da totalidade dos dados de excreção reportados concluiu-se que a conjugação de AA com o GSH ultrapassa a formação do metabolito reactivo GA, existindo uma biodisponibilidade de AA nos humanos de cerca de 20-49%. Esta é metabolizada a GA que por sua vez também é destoxificada pela conjugação com o GSH (Fuhr et al., 2006). Assim, a exposição interna relativa à GA proveniente da dieta humana é pelo menos duas a quatro vezes menor em relação aos ratos e ratinhos, respectivamente (Fennell and Friedman, 2005; Boettcher et al., 2006b; Fuhr et al., 2006; Gargas et al., 2009). Do ponto de vista da avaliação do risco, este facto é bastante importante porque, em seres humanos, haverá menos produtos das reacções electrofílicas do metabolito reactivo.
A AA e a GA são igualmente e uniformemente distribuídas em todos os tecidos e têm um tempo de semi-vida de cerca de 5h no soro de roedores (Doerge et al., 2005b; Doerge et al., 2005c). No entanto, a conversão de AA a GA no modelo de rato parece
- 20 -
ser saturada, verificando-se uma diminuição dose-dependente na oxidação da AA quando a dose é aumentada de 3 mg/Kg para 50 mg/Kg (Kirman et al., 2003; Gargas et
al., 2009). Modelos toxicocinéticos de base fisiológicas para a AA no rato sugerem que
a oxidação de AA pelo CYP2E1 fica saturada com doses superiores a 10 mg/Kg pc/dia (Kirman et al., 2003). Como as exposições no homem não foram realizadas com doses superiores a 3 mg/Kg, não existem comparações directas com o rato. Assim, na ausência de evidência directa, a constatação da dependência da dose em ratos indica também uma probabilidade de ocorrência de dependência da dose nos seres humanos (Gargas et al., 2009).
Alterações na conversão de AA a GA também foram estudadas por Doerge e colaboradores nos seus estudos toxicocinéticos de AA e GA no soro e nos tecidos de ratinhos B6C3F1 (Doerge et al., 2005b) e em ratos Fischer 344 (Doerge et al., 2005c) após administração aguda de 0,1 mg/Kg pc de AA por diferentes vias. Estes autores observaram nos dois estudos uma atenuação da biodisponibilidade de AA pela via oral, em média entre 30-40% em comparação com a via intravenosa. No entanto, verificaram um efeito de primeira passagem ou outra alteração cinética que resultou num aumento da exposição interna relativa à GA (2-7 vezes superior). Um efeito similar na exposição à GA ocorreu quando a dose administrada foi reduzida, o que sugere que para menores taxas administradas, a conversão de AA em GA é mais eficiente.
Outros factores que parecem ser importantes na cinética de formação de GA nos seres humanos são os designados factores externos, como o consumo de álcool e hábito tabágico, idade e sexo (Dybing et al., 2008; Vesper et al., 2008; Vikström et al., 2010). No caso particular do consumo de álcool, alguns autores referem que este favorece a metabolização de AA ao epóxido reactivo por activação do CYP2E1 (Dybing et al., 2008). No entanto, recentemente, num estudo populacional, a associação do consumo de álcool mostrou diminuir os aductos de GA o que é explicado pelos autores como um possível efeito competitivo ao nível do substrato entre o etanol e a AA para o CYP2E1 (Vikström et al., 2010). Neste sentido, os aductos de GA-Hb foram significativamente menores em indivíduos que consumiram maiores quantidades de álcool (> 14,4 g de etanol/dia), relativamente aos indivíduos que consumiram menores quantidades de álcool (< 14,4 g de etanol/dia) (Vesper et al., 2008).
Eventualmente, a dependência metabólica da AA em GA pelo CYP2E1 será de extrema importância em situações de polimorfismos genéticos para o CYP2E1 e consequente variabilidade na actividade enzimática ou em situações de doença, podendo
- 21 -
assim, originar diferentes susceptibilidades na toxicidade da AA em humanos (Vesper
et al., 2008; Vikström et al., 2010).
O conhecimento sobre o metabolismo da AA é importante para a avaliação da segurança como objectivo global e em particular é necessário para o planeamento das estratégias a seguir nos estudos toxicológicos do composto. A GA demonstrou ser o principal metabolito reactivo tendo um papel importante na cancerigénese em roedores (Rice, 2005). Factores como a menor metabolização da AA em GA nos seres humanos em relação a ratos, em doses similares, sugerem uma proteção adequada nos processos de destoxificação da AA e GA contra a toxicidade das doses da dieta humana (Gargas et
al., 2009). No entanto, no homem a GA mostrou ter uma menor extensão de conjugação
pelo GSH e uma efectiva eliminação via hidrólise, enquanto os ratos hidrolisam GA de forma limitada. Apenas uma compreensão global dos mecanismos de acção envolvidos dissipará as dúvidas levantadas por algumas abordagens toxicológicas mencionadas nas próximas secções.
1.4.
Toxicodinâmica
Ambos os compostos, AA e GA ligam-se covalentemente in vivo com nucleófilos celulares formando diferentes tipos de aductos macromoleculares nos tecidos e na corrente sanguínea (Figura 3) (Calleman et al., 1990; Friedman, 2003; Gamboa da Costa et al., 2003). Nas proteínas, a formação dos aductos ocorre principalmente nos grupos sulfidrilo (SH) e amino N-terminal dos aminoácidos (α-NH2) (Friedman, 2003).
Para a hemoglobina (Hb), o grupo α-NH2 N-terminal da valina parece ser o local
nucleofílico preferencial para a acção electrofílica da AA e GA, em detrimento do grupo SH e NH da cisteína e histidina, originando respectivamente o [N-(2 carbomoiletil)- L-valina] e o [N-(2-carbamoil-2-hidroxietil)-RS-valina)]. Este caso particular, uma vez que o grupo SH é muito mais reactivo, justifica-se com a organização tridimensional dos aminoácidos na Hb (Friedman, 2003).
Os aductos com a Hb representam a concentração ao longo do tempo de AA e GA na circulação sanguínea, durante a vida útil dos eritrócitos (~125 dias) e a concentração dos aductos formados é proporcional à dose interna dos compostos (Bergmark, 1997; Fennell et al., 2005). Vários estudos apresentam níveis de aductos de Hb
- 22 -
significativamente superiores em fumadores do que em não fumadores (Bergmark, 1997; Hagmar et al., 2005). Estas medições dos aductos Hb-AA e Hb-GA são habitualmente efectuadas com o intuito de obter orientações sobre os níveis de exposição (Bergmark et al., 1993; Hagmar et al., 2001; Hagmar et al., 2005; Vesper et
al., 2008). Por exemplo, Bergmark (1997) detectou os seguintes montantes de
carbomoiletil-valina no sangue de seres humanos: 31 pmol/g globina em não fumadores; 54 pmol/g globina em pessoal de laboratório que trabalha com a AA; e 116 pmol/g globina nos fumadores. O montante basal de aductos (não fumadores) parece ser proveniente da dieta e é considerado um biomarcador de dose interna. Observações semelhantes foram efectuadas por outros grupos de investigação (Hagmar et al., 2001; Hartmann et al., 2008).
A formação de aductos com o DNA após exposição à AA também tem sido relatada
in vivo (Segerback et al., 1995; Doerge et al., 2005c). Contudo, a capacidade
mutagénica tem sido sobretudo atribuída ao metabolito reactivo, GA, uma vez que a reacção directa da AA com o DNA in vitro é relativamente lenta (Solomon et al., 1985; Besaratinia and Pfeifer, 2005) e não foi demonstrado que ocorra in vivo (Segerback et
al., 1995). As investigações mostraram que a GA, mas não a AA, dá origem a níveis
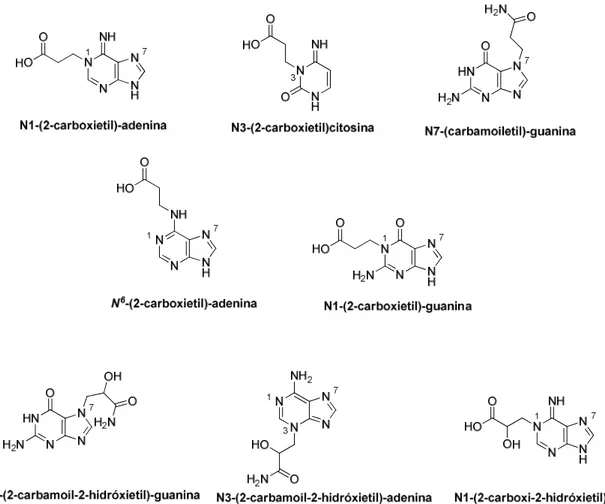
detectáveis de aductos de DNA nos roedores expostos à AA (Segerback et al, 1995). A análise estrutural dos produtos das reacções da AA com o DNA in vitro revelaram a formação dos seguintes aductos alquilados em ordem decrescente de abundância, N1(-2-carboxietil)-adenina, N3-(2 carboxietil) citosina, N7 (carbamoiletil) guanina, N6-(2 carboxietil) -adenina, e N1-(2 carboxietil) guanina (Figura 4) (Solomon et al., 1985).
A GA, por sua vez, forma aductos com as bases púricas do DNA in vitro e in vivo em quantidades apreciáveis (Gamboa da Costa et al., 2003; Besaratinia and Pfeifer, 2007). Esta reacção ocorre rapidamente e tem como resultado predominante a formação do aducto N7-(2-carbamoíl-2-hidroxietil)-guanina (N7-GA-Gua) (Figura 4) (Solomon et
al., 1985; Gamboa da Costa et al., 2003). A formação deste aducto no DNA de ratinhos
tratados com AA ou GA observou-se ser cerca de 100 vezes mais extensa que a formação do aducto derivado da GA N3-(2-carbamoíl-2-hidroxietil)-adenina (N3-GA-Ade), em todos os orgãos testados, ou seja, fígado, pulmão e rim (Gamboa da Costa et
al., 2003). O N1-(2-carboxi-2-hidroxietil)-adenina (N1-GA-Ade) é outro dos aductos de
- 23 -
Figura 4. Estrutura química dos principais aductos de DNA da acrilamida e da glicidamida. N7-(2-carbamoíl-2-hidroxietil)-guanina (N7-GA-Gua) é o aducto de DNA predominante derivado da GA. Adaptado de Besaratinia and Pfeifer (2005).
Alguns autores detectaram e quantificaram os aductos do metabolito reactivo GA, N7-GA-Gua, em múltiplos tecidos de roedores, após uma exposição oral à AA (Segerback et al., 1995; Doerge et al., 2005b; Doerge et al., 2005c). Contudo, os dados sobre aductos de DNA em humanos são escassos (Klaunig, 2008). Enquanto a concentração de AA ligada ao grupo N-terminal da Hb se encontra fortemente relacionada com a exposição à AA, o seu análogo, GA, correlaciona os aductos de GA-DNA e é considerado um biomarcador para a dose genotóxica reflectindo a capacidade individual da activação da AA a GA (Klaunig, 2008).
Vários estudos referem um maior número de aductos nos ratinhos do que nos ratos, o que se correlaciona com a maior conversão metabólica de AA a GA nos ratinhos quando comparado com os ratos (Klaunig, 2008). Doerge e colaboradores em dois estudos efectuados com medição de formação de aductos de GA-DNA no fígado de ratos (Doerge et al., 2005c) e de ratinhos (Doerge et al., 2005b) constataram que as
- 24 -
diferenças de espécies na metabolização de AA e posterior formação de aductos de DNA parecia minimizada numa via oral de exposição, principalmente porque o teor de AA nos alimentos é reduzido.
A reparação da lesão no DNA é outro dos parâmetros toxicodinâmicos que pode representar o passo determinante que conduz a efeitos tóxicos (Gargas et al., 2009). A reparação do DNA seria uma fonte provável para a não linearidade da resposta tumoral da AA (Dybing and Sanner, 2003). As publicações sobre este parâmetro são actualmente ainda muito limitadas.
1.4.1. Espectro de efeitos tóxicos
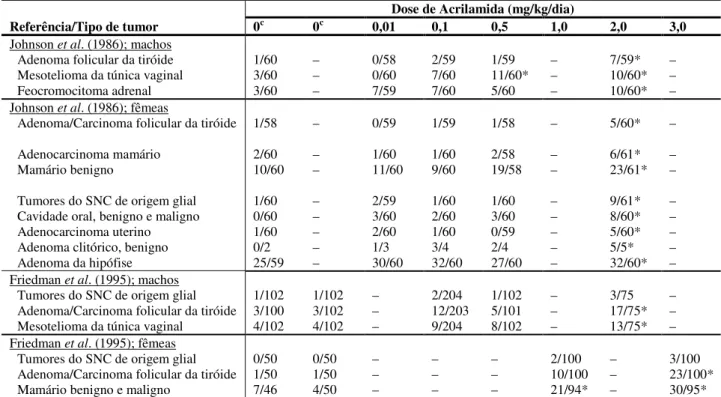
De um modo geral, os principais efeitos tóxicos conhecidos da AA são a neurotoxicidade em humanos e animais, a toxicidade reprodutiva e do desenvolvimento em roedores e genotoxicidade e carcinogenicidade em roedores (Rice, 2005). Na verdade, o potencial cancerígeno da AA em modelo animal é desde há algum tempo conhecido (Johnson et al., 1986; Friedman et al., 1995), o que justifica o renovado interesse no estudo da AA e do seu metabolito reactivo GA desde a descoberta da via alimentar como fonte de exposição.
Desde 1994 que a International Agency for Research on Cancer (IARC, 1994) classificou a AA como um composto provavelmente cancerígeno para o homem (grupo 2A), baseado na constatação de cancerigenicidade em roedores em exposições crónicas e estudos genotóxicos in vitro e in vivo. Contudo, o risco de um processo cancerígeno na população por consumo oral de AA permanece incerto devido aos aparentes resultados discordantes entre os bioensaios de cancro realizados em roedores, que relataram o aparecimento de tumores em vários locais após exposição crónica, e os estudos epidemiológicos que sugerem, mas fornecem pouca ou nenhuma evidência de mortalidade por cancro (ambos os tópicos serão desenvolvidos nos pontos 1.5.2 e 1.5.3)(Klaunig, 2008; Parzefall, 2008), após repetida e prolongada ingestão de AA nos níveis actuais em alimentos e na água da rede pública. Actualmente, o limite experimental usual para a cancerigénese é de 300 µg/Kg pc/dia de AA (BMDL –
- 25 -
margem de segurança humana de 300 vezes, de acordo com o consumo médio diário estimado para a AA (1 µg/Kg pc/dia) (FAO/WHO, 2005).
Em relação à neurotoxicidade e toxicidade reprodutiva, as avaliações de risco são bastante consensuais na afirmação de ausência de risco significativo por via alimentar (FAO/WHO, 2005). A neurotoxicidade da AA tem sido extensivamente estudada em vários modelos experimentais, como ratos, ratinhos, macacos, cães e gatos, por numerosos regimes de dose e duração de administração. Vários sinais neurotóxicos foram encontrados, tais como a fraqueza muscular, ataxia dos membros posteriores e perda da função motora, degeneração dos nervos periféricos e alterações morfológicas e da velocidade de condução nervosa, mas apenas para doses elevadas de AA (Shipp et
al., 2006). No homem, os efeitos neurotóxicos têm sido reconhecidos desde a década de
90, mas essencialmente em relação a uma exposição ocupacional. Intoxicações acidentais e exposições crónicas resultaram em provas de neurotoxicidade em trabalhadores, consistentes com os resultados obtidos em animais (Shipp et al., 2006). Embora tenham sido relatadas principalmente neuropatias periféricas, outras partes do sistema nervoso também mostraram ser afectadas, tendo sido nomeadamente encontrada lesão celular nas células Purkinje do cerebelo e degradação dos axónios distais do sistema nervoso central e sistema nervoso periférico (Shipp et al., 2006; Parzefall, 2008). A degradação dos nervos terminais foi reportada como estando na origem do comprometimento das funções cognitivas e de lesões no córtex cerebral, tálamo e hipocampo (Shipp et al., 2006).
Em alguns estudos, tem sido efectuada a comparação directa do
no-observed-adverse-effect level (NOAEL) para efeitos neurotóxicos em animais com dados de seres
humanos, com base na medida de dose interna de aductos AA-Hb em ambas as espécies (Calleman, 1996). Para as pessoas expostas, o valor de NOAEL extrapolado para a neurotoxicidade foi o de 0,2-0,5 mg/Kg pc/dia e o lowest-observed-adverse effect level (LOAEL) foi o de 2 mg/Kg pc/dia (Exon, 2006; Parzefall, 2008). Estes valores estão muito acima da média de exposição alimentar estimada pela WHO (1 µg/Kg pc/dia) (FAO/WHO, 2005), usualmente utilizada em modelos de avaliação de risco e que fornecem uma margem de segurança de cerca de 200-500 vezes. Embora alguns neurologistas estejam preocupados com o potencial neurotóxico de uma acumulação de AA, a maioria acredita que a exposição de seres humanos a níveis relativamente baixos de AA na dieta não resultará em neuropatia clínica (Exon, 2006).
- 26 -
A toxicidade reprodutiva, também observada em animais de laboratório expostos a dose elevada de AA, mas nunca relatada em seres humanos, demonstrou uma redução de fertilidade, efeitos letais dominantes e efeitos adversos na morfologia e quantidade de espermatozóides (Shipp et al., 2006). Ainda assim, a AA não foi teratogénica em ratos (Tyla et al., 2000; Exon, 2006; Parzefall, 2008). O NOAEL para efeitos na reprodução foi estimado em 2-5 mg/Kg pc/dia (Tyla et al., 2000; Parzefall, 2008), dependendo do endpoint de fertilidade ou morte embrionária. Este valor é, pelo menos, quatro vezes superior ao respectivo parâmetro de neurotoxicidade e 2000 vezes superior à exposição dietética estimada (Dybing and Sanner, 2003). Por isso, foi considerado altamente improvável que qualquer toxicidade reprodutiva em seres humanos seja resultado da exposição a AA na dieta, embora haja algumas preocupações sobre os efeitos cumulativos de baixos níveis por exposição crónica (FAO/WHO, 2005).
Os factores em causa na avaliação de risco da AA perante os aspectos toxicológicos conhecidos e a constatação de uma difundida exposição humana a baixos níveis deste composto através da dieta, prendem-se sobretudo com o renovado interesse no seu metabolito reactivo (GA), principalmente, porque se sabe que os agentes químicos genotóxicos e a maior parte dos agentes cancerígenos humanos são metabolizados a electrófilos reactivos (Jägerstad and Skog, 2005). Este último facto pode, por conseguinte, indicar um potencial genotóxico da AA e um aumento de risco de cancro no homem, visto que a AA tem a capacidade de ter um metabolito (GA) que forma aductos com o DNA (Gamboa da Costa et al., 2003). Além disso, a AA está presente nos alimentos em níveis consideravelmente superiores aos apresentados por outros bem conhecidos cancerígenos alimentares, como hidrocarbonetos policíclicos aromáticos, carbamato de etilo e aminas heterocíclicas (FAO/WHO, 2005; Jägerstad and Skog, 2005; Ferguson and Philpott, 2008).
Para harmonização de conhecimento e para uma análise de risco inequívoca, uma tarefa a realizar é sem dúvida esclarecer a relação dose-resposta para baixas doses de AA e os mecanismos de toxicidade associados, principalmente quando está em causa um possível mecanismo genotóxico (acção de GA) sem limiar de efeito tóxico. Por outro lado, deve considerar-se que a AA é apenas um entre muitos compostos electrofílicos observados a ocorrer com uma exposição de base natural (aductos de AA-Hb).
- 27 -
1.5.
Acrilamida como provável cancerígeno humano
1.5.1. Genotoxicidade
A avaliação do IARC, em 1994, de que a AA é um provável cancerígeno humano dependeu, em parte, de evidências de que a AA induzia mutações genéticas e aberrações cromossómicas em células germinativas e em células somáticas de roedores in vivo e de que induzia mutações genéticas e aberrações cromossómicas em culturas de células in
vitro (IARC, 1994). Estudos mais recentes têm avaliado extensivamente a toxicidade
genética da AA, bem como do seu epóxido reactivo, in vitro e in vivo, confirmando e ampliando as evidências anteriores de que a AA é genotóxica e de que a sua genotoxicidade in vivo é essencialmente mediada por biotransformação a GA (Besaratinia and Pfeifer, 2004; Besaratinia and Pfeifer, 2005; Carere, 2006).
Alguns autores avaliaram o potencial mutagénico da AA e de GA em sistemas bacterianos (Hashimoto and Tanii, 1985; Tsuda et al., 1993) e em células de mamífero (Besaratinia and Pfeifer, 2003; Koyama et al., 2006; Mei et al., 2008). Outros estudos abordaram os potenciais efeitos genotóxicos destes compostos, pela avaliação de alterações cromossómicas estruturais (Adler et al., 1988; Tsuda et al., 1993; Manjanatha
et al., 2006; Martins et al., 2007), lesões do DNA, ensaios de reparação de DNA
(Butterworth et al., 1992; Tsuda et al., 1993; Baum et al., 2005; Hansen et al., 2010), ou em ensaios de transformação celular (Tsuda et al., 1993; Park et al., 2002a). De uma forma geral, as observações resultantes encontram-se descritas nos tópicos seguintes.
1.5.1.1. Mutações genéticas em células procariotas
Apesar de a AA não ter induzido mutações genéticas nos ensaios bacterianos (teste de Ames) em diferentes estirpes de Salmonella typhimurium e Escherichia coli, na presença ou ausência de um sistema de activação metabólica exógeno de mamíferos (Hashimoto and Tanii, 1985; Tsuda et al., 1993), os resultados de outros testes de mutagenicidade in vitro e in vivo foram predominantemente positivos a fornecer elementos de prova para o potencial cancerígeno humano da AA (Besaratinia and Pfeifer, 2005). A GA, por sua vez, foi mutagénica no teste de Ames mesmo sem activação metabólica (Hashimoto and Tanii, 1985), o que levou alguns autores a